BORE
Le bore est l’élément chimique de symbole B et de numéro atomique Z = 5. Bien que sa chimie soit mal connue et en pleine évolution, les emplois de ses dérivés sont anciens, nombreux et importants.
Au début du Moyen Âge, on importait en Europe du borax, venu des Indes, sous des noms (d’origine arabe?) tels que «tincar», «tinckal»... Vers 1500, ce produit est appelé (origine perse) «baurach», «bauraq»... Extrait du borax en 1702 par Homberg et d’abord nommé «sel sédatif», «sel volatil narcotique de vitriol», «acide du borax», «acide boracin», «acide boracique», l’acide borique a été ainsi appelé, vers 1810, par Gay-Lussac et Thénard. Le mot bore est dû aux mêmes auteurs qui, en même temps que Davy, et de façon indépendante, ont préparé pour la première fois, en 1808, du bore élémentaire très impur. Les alchimistes se servaient déjà du borax comme flux dans la fusion et la soudure des métaux. Encore aujourd’hui, le borax et l’acide borique entrent dans la composition des flux utilisés en métallurgie; ils servent à l’ignifugation du bois et des tissus; on les trouve dans la composition des émaux utilisés en céramique et dans la composition de verres du type Pyrex; on les emploie en galvanoplastie, photographie, tannerie, papeterie, blanchisserie, teinturerie et pour la fabrication des colles, des condensateurs électrolytiques... L’acide borique, qui figure comme antiseptique dans la pharmacopée, est toxique; il ne doit pas être employé pour la conservation des aliments. De son côté, la borocalcite est utilisée comme fertilisant (betterave, pommier...). Les perborates servent au blanchiment des textiles. Le fluorure de bore est un catalyseur énergique, analogue au chlorure d’aluminium, utilisé en chimie organique. Les hydrures, proposés comme carburants pour fusées, servent au «dopage» des semi-conducteurs, et les borohydrures (qui permettent d’obtenir facilement les hydrures) sont des réducteurs sélectifs utilisés en chimie organique. Il faut encore citer le nitrure de bore utilisé comme réfractaire, le carbure de bore et les borures des métaux de transition employés comme réfractaires ou comme abrasifs, le borure de fer permettant de désoxyder l’acier et d’en améliorer les propriétés mécaniques. Ces corps servent aussi dans l’industrie nucléaire pour absorber les neutrons lents (10B ayant une section efficace de capture cinq fois plus grande que celle du 11B); ils semblent devoir servir à renforcer les matières plastiques plus efficacement que les fibres de verre. L’emploi du bore élémentaire a été préconisé en particulier pour améliorer les caractéristiques des combustibles pour fusées.
Le bore a pour masse atomique 10,82. Il possède deux isotopes naturels, 10B (18,8 p. 100) et 11B (81,2 p. 100); on sait, de plus, obtenir deux isotopes radioactifs de périodes courtes, 8B et 12B, par bombardement de 9Be, 10B, 11B, 12C et 15N.
Sa structure électronique est 1s 2, 2s 2, 2p 1; il appartient de ce fait au groupe III b de la classification périodique (B, Al, Ga, In, Tl). D’après cette structure, seul l’électron 2p n’étant pas apparié, le bore devrait être monovalent; en fait il suffit de fournir une énergie d’activation très faible (167 joules par atome-gramme) pour que, l’un des électrons 2s passant sur l’orbitale 2p , le bore soit toujours trivalent. La différence d’énergie entre les niveaux 2s et 2p est trop faible pour qu’il puisse être bivalent; ceci cadre du reste avec le fait que le bore lui-même et ses dérivés sont diamagnétiques et avec le fait que dans des corps tels que (BX2)2, (B(OH)2)2 ou (BO)2 on trouve des structures fragiles du type 礪 B–B 麗, et, dans (BX)n , des structures spatiales dans lesquelles le bore est trivalent.
Suivant une règle générale, en tant que premier élément d’une colonne de la classification, le bore se différencie très nettement des éléments qui le suivent. En particulier, il ne donne pas d’ion positif, car, à cause de son petit diamètre atomique et du faible effet d’écran, ses électrons sont très fortement attirés par le noyau: ceci se traduit par une énergie d’ionisation environ 1,3 fois plus grande que celle des autres éléments de sa famille.
Ne pouvant établir que trois covalences, le bore sera entouré dans ses composés, non pas d’un octet, mais seulement de six électrons, et la chimie du bore est caractérisée par ce déficit électronique.
À l’état élémentaire, le bore se cristallise dans des systèmes compacts (rhomboédrique ou quadratique) très stables et favorisant au maximum la mise en commun des électrons, d’où sa dureté et son point de fusion élevé. Les atomes de bore se groupent alors en triangles équilatéraux, lesquels s’associent entre eux en mettant leurs côtés en commun; on arrive ainsi à l’octaèdre ou à l’icosaèdre (fig. 1). Quoi qu’il en soit, il n’existe pas, dans ces configurations, un ensemble de covalences normales (dans l’icosaèdre, par exemple, douze atomes de bore échangent trente liaisons au moyen seulement de trente-six électrons); les mêmes considérations sont valables pour les borures métalliques, ou pour le carbure de bore.
Lorsqu’un atome de bore tricovalent est engagé dans une molécule, il va, de façon à compléter son octet, servir d’accepteur à une molécule ou à un ion contenant un atome (O, S, N...) pouvant servir de donneur. Ceci provoque soit la polymérisation fréquente des dérivés du bore, soit la formation de composés d’addition, et, lorsque l’atome donneur se trouve dans une chaîne liée au bore, tout en étant séparé par quatre ou cinq autres atomes, la formation de chélates.
Ce déficit électronique est à l’origine de la structure très particulière des hydrures de bore (pont hydrogène).
Enfin, toujours pour la même raison, en se combinant avec des éléments excédentaires en électrons, le bore donne des composés hétéro- ou polycycliques.
1. État naturel
Le bore entre pour 0,001 p. 100 dans la composition de la croûte terrestre où il est très disséminé. C’est un oligo-élément nécessaire au développement normal des animaux et des végétaux.
Le bore est trop réactif pour exister à l’état libre, on le trouve essentiellement sous forme oxydée dans environ soixante minéraux constitués par des borates associés à des silicates, phosphates, aluminates, sulfates, fluorures, chlorures..., et dont les gisements sont localisés en Amérique (Californie, Chili), en Asie centrale et en Europe (U.R.S.S., Allemagne, Italie); la France ne possède aucun gisement.
Après extraction, ces minéraux sont mis en solution par l’acide sulfurique ou par le carbonate de soude. On précipite les impuretés et on laisse cristalliser, suivant le cas, l’acide borique ou le borax, ce dernier pouvant être obtenu par simple recristallisation de la kernite. L’acide borique et le borax sont les deux produits commerciaux les plus importants.
On trouve également des produits boraciques dans certains lacs salés (Searles Lake en Californie). On récupère en Toscane l’acide borique abandonné par des jets de vapeur d’eau surchauffée (100-215 0C) d’origine volcanique qui sont exploités comme sources d’énergie; la vapeur, hydrolysant des borates dans les profondeurs du sol, contient de l’acide borique et divers sels minéraux. La vapeur qui s’échappe librement des fissures du sol (soffioni ) est simplement condensée dans des bassins (lagoni ).
2. Bore élémentaire
L’obtention de bore de pureté élevée a donné lieu à de nombreux travaux car des traces de corps étrangers agissent considérablement sur son état cristallin et sur ses propriétés physiques et chimiques. Sa préparation est rendue délicate par sa grande réactivité chimique.
Le bore est obtenu par réduction à chaud de l’anhydride borique B23 par un métal très réducteur (Mg, Al, Ca, alcalins), ou par électrolyse à 1 100 0C d’anhydride borique ou de borates fondus contenant en dissolution divers sels de magnésium; il y a alors, en fait, réduction de l’anhydride borique ou des borates par le magnésium libéré lors de l’électrolyse. Le bore ainsi obtenu contient des sels métalliques, des borures qu’on élimine par ébullition avec des acides, puis, éventuellement, par fusion avec B23 et par chauffage sous vide. On arrive ainsi à un produit dont le titre ne dépasse pas 90 p. 100.
Les halogénures de bore (surtout le chlorure) peuvent être réduits par l’hydrogène (procédé Van Arkel ou dérivés) ou par divers métaux (Mg, Al, Fe, Ni, alcalins, Zn...). Le bore ainsi obtenu est de pureté élevée, puisque, selon les conditions opératoires, les produits de la réaction sont éliminés.
On fait appel également à la dissociation thermique des halogénures, du sulfure et surtout des hydrures.
Le bore se présente sous forme d’une poudre brun-noir, couleur qu’il garde après avoir été fondu ou dispersé à l’état de suspension colloïdale.
Ses propriétés physiques semblent varier avec son état cristallin. Il existe en effet, au moins sous deux formes: rhomboédrique 見 stable en dessous de 1 500 0C (douze atomes par maille) et rhomboédrique 廓 stable à haute température (cent huit atomes par maille); on admet au moins une forme quadratique (cinquante atomes par maille) obtenue par refroidissement brutal à partir de 1 500 0C.
Comme conséquence de sa structure électronique, la densité du bore est élevée (2,3); sa dureté est presque aussi grande que celle du diamant; ses coefficients de dilatation et de compressibilité sont très faibles; son point de fusion est élevé (face=F0019 力 2 400 0C) et son spectre de flamme est simple (vingt-cinq raies surtout dans le vert et le jaune).
À cause de sa faible masse atomique, le bore ne vérifie la loi de Dulong et Petit qu’à température élevée.
De même que les propriétés physiques, les propriétés chimiques du bore restent à préciser, car elles ont été le plus souvent déterminées sur des échantillons de pureté insuffisante.
Le bore est capable, éventuellement à température élevée, de s’unir à presque tous les autres éléments, ceci le plus souvent de façon très exothermique. Réducteur extrêmement énergique, il peut agir facilement sur l’ensemble des composés oxygénés, sulfurés, halogénés, azotés... des autres éléments. Sa chimie, dont l’étude est, de ce fait, très délicate, est, par divers aspects, à rapprocher de celles du carbone et du silicium.
L’étude est encore compliquée par des difficultés analytiques. La mise en solution du bore et de certains de ses dérivés est très laborieuse, les séparations souvent complexes et il est délicat de différencier dans un mélange le bore libre du bore combiné (oxydation sélective du bore libre par Ce4+ Mn4-...).
Si on décèle facilement sa présence par le fait qu’il colore la flamme en vert (après transformation éventuelle en halogénure ou en ester de l’acide borique), on dose péniblement le bore par gravimétrie (après séparation à l’état de borate de méthyle) ou mieux par acidimétrie (après transformation en acide borique); pour les traces, on opère par spectrographie, par colorimétrie, ou en mettant en jeu des réactions colorées (papier au curcuma). La précision des dosages est faible (1 à 2 p. 100).
Il est à noter que la nomenclature des dérivés du bore est mal définie.
3. Borures
Le bore réagit sur presque tous les métaux pour donner des borures, corps qu’on sait également obtenir en réduisant l’oxyde du métal par le bore, ou en réduisant un composé du bore (halogénure, borate...) par un métal ou par un composé métallique (hydrure, carbure...). On peut aussi électrolyser un borate fondu contenant en dissolution un dérivé métallique (oxyde, fluorure...). On a décrit de nombreux borures simples ou mixtes – certains métaux donnant jusqu’à six borures – confirmés par analyse thermique ou radiocristallographique. Ces corps sont bien cristallisés et ont l’éclat métallique; ils sont à rapprocher des carbures, des nitrures ou même des alliages métalliques.
On classe les borures d’après leur constitution cristallographique, suivant qu’ils contiennent des atomes de bore isolés (M2B), groupés par paires (M3B2), ou disposés en chaînes linéaires simples (MB) ou doubles (M3B4); ces atomes peuvent se grouper en cycles hexagonaux plans (MB2), en réseaux octaédriques (MB6) ou icosaédriques (MB12). Ces structures expliquent que les borures soient généralement durs, à haut point de fusion, chimiquement plus inertes et électriquement plus résistants que les métaux correspondants; quelques-uns sont presque les plus durs, les plus réfractaires de toutes les substances connues et leurs applications sont fondées sur ces caractéristiques extrêmes.
4. Dérivés hydrogénés ou boranes
Le bore et l’hydrogène donnent une série de composés qui sont, suivant le poids moléculaire, gazeux, liquides ou solides.
L’étude de ces produits réducteurs et le plus souvent hydrolysables est particulièrement complexe. Ils sont instables: les hydrures légers se transforment avec perte d’hydrogène en un mélange de termes supérieurs pour aboutir à des solides (BH)n qui n’ont pu être encore étudiés; ces transformations se font à la suite de mécanismes mal connus au cours desquels on passe par le groupe BH3 non isolé. De plus, ces corps sont hautement toxiques et certains d’entre eux forment avec l’air des mélanges auto-explosifs.
Préparés initialement par pyrolyse de tétraborane B4H10 (obtenu lui-même par hydrolyse du borure de magnésium), les boranes s’obtiennent par décomposition thermique du diborane B2H6. Le diborane peut être obtenu à partir des organoboranes ou par action d’un acide de Lewis sur un borohydrure (ou hydroborure, ou boranate), par exemple:

De façon peu satisfaisante, on classe les boranes en deux séries: Bn Hn+4 (B2H6; B5H9; B6H10; B10H14) et Bn Hn+6 (B4H10; B5H11; B7H13; B9H17). En effet, d’une part certains hydrures ne font pas partie de ces groupes, d’autre part, au cours des réactions chimiques, B4H10 se coupe en BH3 et B3H7 alors que B9H15 semble correspondre à l’association, avec perte d’hydrogène, de B4H10 et de B5H9. Enfin les ions borohydrures (BH4)- et (B3H8)- dérivent des hydrures BH3 et B3H7, non isolés, mais qu’on sait stabiliser sous forme de dérivés de coordination tels que R2OB3H7.
La structure des hydrures de bore est très complexe (fig. 2). On admet, par exemple pour B2H6, que deux groupes, BH3 sont liés par «pont hydrogène», c’est-à-dire par une combinaison de l’orbitale de l’hydrogène et des orbitales hybrides du bore; ce type de liaison se retrouve dans les boranes supérieurs. En effet le diborane B2H6 est très différent de l’éthane C2H6 avec lequel il avait été initialement comparé puisque, le bore étant trivalent, il est impossible d’écrire, par exemple, H3B 漣BH3. De plus, les déterminations physico-chimiques montrent que la distance des deux atomes de bore dans B2H6 est supérieure à celle qui existe dans une covalence normale B 漣B. Par ailleurs, bien qu’il existe des dérivés B2H5X, on ne sait obtenir que des corps de formules B2R4H2; ceci implique pour deux atomes d’hydrogène des propriétés particulières. On constate encore que, comme pour une liaison éthylénique, la libre rotation autour de l’axe B 漣B est impossible. Enfin, sous l’action de corps possédant un atome donneur d’électrons (O, N, P...): alcools, éthers, aldéhydes..., le diborane se coupe en donnant des composés d’addition tels que:

La structure des hydrures supérieurs correspond à l’assemblage triangulaire d’atomes de bore, les triangles se groupant entre eux et tendant à former à la limite un icosaèdre. Toutefois l’hydrure B12H12 est inconnu, car sa formation correspondrait à un déficit électronique. Par contre, on connaît l’ion (B12H12)2-, dernier terme de la série (Bn Hn )3-, n étant compris entre 3 et 12, et divers dérivés tels que (B12Cl12)2-. Dans ces conditions pour combler le déficit électronique des carbures neutres qui correspondraient aux ions (Bn Hn )2-, il est possible de remplacer deux atomes de bore par deux atomes de carbone. On passe ainsi à la série des «carboranes» C2Bn Hn+2 , n étant compris entre 2 et 10. Seuls les termes de rang élevé sont relativement bien connus et, fonction des isoméries possibles, la nomenclature reste à établir.
Les borohydrures, eux, sont préparés par action d’un hydrure alcalin (ou du métal en présence d’hydrogène) sur un halogénure de bore:

Ces corps sont des réducteurs très utilisés en chimie organique:

5. Dérivés oxygénés
Il existe une série de sous-oxydes, difficiles à préparer, par exemple en oxydant le bore à température très élevée, et dont les formules seraient comprises entre B23 et B7O; on peut admettre l’existence en particulier de (BO)n et d’un oxyde de formule controversée B6O, B7O ou B132.
Les structures de ces corps sont mal connues et certains donnent par action de l’eau des acides réducteurs (acides hypoboriques), par exemple: H4B24; H3B2; H2B46; H4B22; H2B22..., acides dont on connaît des sels et des esters.
L’anhydride borique B23 est l’oxyde normal qu’on obtient par déshydratation de l’acide borique. Il se présente soit à l’état cristallisé (réseau hexagonal), forme difficilement obtenue par un long recuit à 250 0C, soit à l’état vitreux. La forme vitreuse, très dure, est constituée par l’assemblage de polygones B46, dans lesquels les liaisons B 漣O sont très lâches. À l’état liquide l’anhydride borique est associé; ceci permet d’en expliquer les propriétés physiques (bas point de «fusion», viscosité élevée...).
L’anhydride borique fondu dissout pratiquement tous les oxydes métalliques; par refroidissement de telles solutions, on obtient, soit un verre homogène, soit un dépôt d’oxydes ou de borates. Il est possible, comme dans l’eau, d’obtenir, dans l’anhydride borique fondu, des réactions de double décomposition entre deux sels dissous ou, par électrolyse, un dépôt de bore, de métal ou de borures.
En fonction de sa chaleur de formation élevée (300 kcal/mole) l’anhydride borique n’est réduit que par des réducteurs puissants (alcalins, Mg, Ca...), avec libération de bore et formation de borures.
Acides boriques
Par hydratation, l’anhydride borique donne successivement l’acide métaborique HB2 (monohydrate de B23), puis l’acide orthoborique H3B3 (trihydrate de B23), mais ne semble pas donner l’acide pyroborique H2B47 dont l’existence prête à discussion.
L’acide métaborique (ou monoborique) HB2 existe sous trois formes cristallines; la variété courante, orthorhombique, est constituée par des groupes B3 liés entre eux par liaison hydrogène et disposés en couches parallèles. Il semble exister en solution sous forme dimère:

L’acide orthoborique (ou borique) H3B3 cristallise en paillettes tricliniques incolores ou blanches, onctueuses, qui se disposent en couches parallèles, liées par des liaisons de Van der Waals assez faibles.
La solubilité de l’acide orthoborique dans l’eau est sept fois plus grande à 100 0C qu’à la température ambiante (50 g/l), ce qui permet sa purification. Il est entraîné par la vapeur d’eau, bien avant l’ébullition, peut-être à l’état d’acide métaborique.
En solution l’acide orthoborique est un monoacide très faible qui se dissocie légèrement suivant:

(d’où l’emploi de ses solutions avec du borax comme solutions tampons). Suivant la température et la concentration, il y a simultanément solvatation et condensation avec élimination d’eau des molécules H3B3 et des ions H2B3-, avec formation d’ions tels que [B(OH)4]-, et peut-être même d’ions B2-, (BO3)3-, B472- ...
Avec les composés minéraux, l’acide borique donne des borates; avec les alcools, il donne des esters B(OR)3 très hydrolysables, lesquels, par action des halogénures de bore, se transforment en esters halogénoboriques BX(OR)2 et BX2OR.
Avec les corps polyhydroxylés (glycérine, acides-alcools, acides-phénols...) l’acide borique donne de nombreux complexes. En particulier, la glycérine et surtout le mannitol conduisent à la formation de complexes d’addition qui sont des acides beaucoup plus forts que l’acide borique pur; avec cet acide très faible, on n’observe pas, lors du dosage par la soude, de cassure nette de la courbe de neutralisation, alors que le dosage acidimétrique en présence de mannite est très aisé.
Borates
Par action de la solution d’acide borique sur divers sels métalliques (oxydes, hydroxydes, carbonates...), on obtient de très nombreux borates dont la composition varie avec la température et la concentration, en particulier par hydrolyse ou par changement du degré d’oxydation. La nomenclature de ces sels (qu’on sait également obtenir par voie sèche) étant délicate, car les structures sont incertaines, on écrit très souvent la formule des borates de façon «dualistique»: x B23, y M2O, z H2O (1 諒 x 諒 5, 1 諒 y 諒 3). Les molécules d’eau qui figurent dans de telles formules sont soit de constitution, soit d’hydratation, et peuvent même apparaître, en cours de chauffage, par réaction entre des groupes OH- existant dans le sel; ainsi le borax possède deux molécules d’eau de constitution et huit d’hydratation. On admet volontiers qu’il existe dans les borates, à l’état solide, ou en solution, soit des atomes de bore tétracoordinés, soit des groupes (HO)2 漣B 漣B 漣(OH)2, soit divers ions de formule générale (Bm On )x- , par exemple (BO2)-, (B47)2-, (B58)- de valences et de constitutions mal précisées. On trouve cependant de façon constante, dans les produits solides, le groupe B3 qui peut soit donner des chaînes, soit se cycliser (fig. 3).
Par ailleurs, la solution d’acide borique donne, par action sur les tungstates, les vanadates..., toute une série de complexes, borotungstates, borovanadates..., tels que:

on obtient de même des hétérocomplexes avec les phosphates et les arséniates.
Perborates
Par addition d’eau oxygénée ou de peroxyde de sodium à une solution de borate, ou par électrolyse d’une solution contenant un borate et du carbonate de soude (l’électrolyse produisant de l’eau oxygénée au sein de la solution) on obtient des perborates dont on ne connaît de façon certaine qu’un petit nombre.
Il faut distinguer les peroxyhydrates constitués par addition d’eau oxygénée sur un borate (BO2Na, H22, 3H2O) et les peroxyborates, plus rares, qui comportent une liaison peroxyde sur le bore (BO3Na).
À l’état solide, les perborates sont peu stables, ils se décomposent à chaud en borate et oxygène, la décomposition des produits très riches en oxygène pouvant être explosive.
Peu solubles dans l’eau, les perborates s’hydrolysent avec libération d’eau oxygénée, c’est ce qui fait utiliser certains d’entre eux (BO3Na, 4H2O) pour le blanchiment des textiles.
6. Dérivés halogénés
Les dérivés normaux BX3, de formation très exothermique, sont très stables. On a identifié, mais non isolé, des dérivés mixtes (par exemple BF2Cl...). On connaît, de plus des halogénures B2X4, peu stables, obtenus en particulier par décharge électrique sous basse pression dans BX3 et qui se décomposent en donnant (BX)n .
Les halogénures BX3 s’obtiennent, de façon classique, par action de l’halogène ou d’un dérivé halogéné sur du bore, un dérivé du bore ou sur un mélange d’anhydride borique et de charbon.
Le fluorure de bore est préparé en particulier par action sur l’anhydride borique, du fluoborate de potassium, de la cryolite ou d’un mélange de fluorure de calcium et d’acide sulfurique, mélange qui libère de l’acide fluorhydrique.
La molécule des halogénures de bore est plane, le bore étant au centre d’un triangle équilatéral. N’étant pas dipolaires, ces corps ne sont pas ionisants.
Alors que les autres halogénures s’hydrolysent de façon brutale, le fluorure de bore est très soluble dans l’eau; il donne en particulier de l’acide fluoborique H(BF4):

c’est un acide fort qu’on prépare, en fait, par action de l’acide borique sur l’acide fluorhydrique; on en connaît de nombreux sels minéraux simples ou complexes (fluoborates ou bore-tétrafluorures) et de nombreux dérivés organiques. L’ion (BF4)- est tétraédrique.
Les halogénures de bore donnent des composés de Van der Waals relativement peu stables et surtout, par suite de leur déficit électronique, des composés de coordination avec des molécules ou des ions contenant un atome donneur (O, S, N, P, X...). Suivant le cas, ces combinaisons sont plus ou moins dissociables à chaud et plus ou moins hydrolysables. Ceci entraîne que le fluorure de bore (et à un degré moindre le chlorure) est, soit à l’état libre, soit à l’état de combinaisons, un catalyseur extrêmement important en chimie organique, car il est doué d’une activité exceptionnelle, très supérieure à celle du chlorure d’aluminium. Il est utilisé dans des réactions d’addition, d’isomérisation, de polymérisation, d’alcoylation..., puisque, pratiquement, il se combine à toutes les fonctions organiques.
7. Dérivés azotés
Parmi les dérivés azotés: amides B(NR2)3, amidure B(NH2)3, imidure B2(NH)3, cyanure B(CN)3, isocyanate B(OCN)3..., le nitrure de bore BN est le plus intéressant. On le prépare par action d’un dérivé azoté (NH3, NH4, Cl, NO...) sur le bore, le chlorure de bore ou sur un mélange capable de libérer du bore.
C’est une poudre blanche qui s’oxyde à l’air en dessous de 1 000 0C et qui cristallise dans le même système que le graphite, mais qui, à cause de sa structure électronique, et, contrairement à ce dernier, n’est pas conducteur du courant électrique. Il est employé comme réfractaire.
On observe, avec le nitrure de bore, une allotropie analogue à l’allotropie graphite-diamant. En soumettant la forme hexagonale (BN)3 à des températures et à des pressions élevées (1 800 0C, 8 500 bar), les cycles hexagonaux gauchissent; on obtient une forme ayant la structure du diamant, qui, comme lui-même, est instable à température élevée sous la pression ordinaire (fig. 4). Le produit obtenu appelé borazon a une dureté minéralogique supérieure à celle du diamant.
8. Carbure de bore
La formule B12C3 est encore discutée à cause des difficultés de dosage des deux éléments (libres ou combinés) et elle est rendue incertaine par la formation de solutions solides de substitution (remplacement, dans la maille, d’atomes de carbone par du bore).
Il est obtenu à température élevée soit par synthèse directe, soit en faisant réagir du carbone sur de l’anhydride borique ou en chauffant du bore dans un mélange d’oxyde de carbone et d’hydrogène.
En raison de sa structure icosaédrique, le carbure est plus dur que le diamant, il est relativement peu fragile et son point de fusion est élevé (2 350 0C). Il est extrêmement réducteur. On l’emploie comme abrasif (analogue au carbure de tungstène), comme réfractaire, comme désoxydant en métallurgie et, en physique nucléaire, comme matériau absorbant les neutrons lents.
9. Dérivés organiques
D’une part, il existe de très nombreux composés de coordination entre un dérivé du bore, le bore servant d’accepteur, et des molécules organiques possédant un atome donneur (O, S, N...). D’autre part, on connaît des dérivés organoboriques dans lesquels le carbone d’un radical organique est lié directement par covalence à un atome de bore, ce qui confère à ces corps des propriétés spécifiques.
Extrêmement nombreux sont les composés d’addition entre un dérivé de bore et une molécule contenant un atome donneur (alcools, phénols, acides, éthers, aldéhydes, cétones...), ils sont du type R2OBX3 ou même R2O(BF3)2 et évoluent souvent avec formation d’ions complexes, où le bore est tétracoordiné:
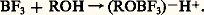
Avec des corps possédant deux fonctions (polyols, diphénols...), on obtient, avec un mécanisme semblable, des dérivés chélatés lorsque la chaîne organique dont fait partie l’atome donneur comporte au moins trois ou quatre atomes le séparant du bore. On obtient ainsi des cycles hexa- ou pentagonaux, dans lesquels le bore est tétracoordiné (fig. 5).
Les composés organoboriques BR3, R étant aliphatique ou aromatique, sont d’étude délicate, car ils sont en général relativement peu stables et parfois auto-inflammables.
On les prépare, en particulier, par action d’un organométallique (Al, Mg, Zn) sur l’anhydride borique, un halogénure de bore ou un borate organique...:
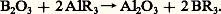
En plus des dérivés simples BR3, on connaît, d’une part, des dérivés mixtes BR2R et BRR R , assez instables sauf lorsque le bore est lié par deux covalences B 漣C à un même radical

et, d’autre part, des dérivés instables BR2X et BRX2.
Par oxydation ménagée de BR3, on obtient des corps instables, souvent auto-inflammables et mal étudiés tels que BR2OH ou BR(OH)2, éthers de l’acide boronique.
Avec des métaux très électropositifs, des composés de type salin ou des anions, les organoboriques conduisent à des ions complexes:

Enfin, pour compléter l’octet du bore, les organoboriques donnent de nombreux composés d’addition avec des molécules contenant un atome donneur, par exemple R3NBR3.
10. Hétérocycles
En faisant réagir un dérivé du bore (hydrogéné, halogéné ou organique) sur un dérivé de l’oxygène, du soufre, du carbone ou de l’azote, on obtient des hétérocycles plans, octogonaux ou pentagonaux, et le plus souvent hexagonaux, ainsi que des dérivés polycycliques (fig. 6). Par exemple:

Le borazole B33H6 est bien connu. C’est un liquide stable semblable au benzène, dont il est isostère et isoélectronique [cf. AROMATICITÉ], et dont on connaît de nombreux substitués sur l’azote ou sur le bore.
Le boroxol et le borsulfol (qui n’existent que si le bore porte des substituants) sont à considérer comme les trimères de dérivés de l’acide métaborique HB2 et de l’acide métathioborique HBS2.
bore [ bɔr ] n. m.
• 1809; de borax
♦ Chim. Élément atomique (B; no at. 5; m. at. 11) du même groupe que l'aluminium, le gallium et l'indium. Halogénure, nitrure de bore.
⊗ HOM. Bord, bort.
● bore nom masculin (de borax) Corps simple présentant avec le carbone certaines analogies. (Élément chimique de symbole B.) Numéro atomique : 5 Masse atomique : 10,81 Température de fusion : ∼ 2 300 °C Masse volumique : 2,34 g°cm3 ● bore (homonymes) nom masculin (de borax) bord nom masculin bort nom masculin
bore
n. m. CHIM élément non métallique (B), de numéro atomique Z = 5, utilisé comme élément d'addition dans les aciers pour améliorer certaines de leurs propriétés.
⇒BORE, subst. masc.
CHIM. Corps simple, métalloïde dur et noirâtre, se rapprochant du carbone. Acier au bore (GOLDSCHMIDT, L'Aventure atomique, 1962, p. 210).
Rem. Boro- entre comme 1er élément de compos. dans la formation de subst. (et des adj. correspondants) : borosilicate, subst. masc. ,,Sel double formé de la combinaison d'un borate et d'un silicate : ce verre [le pyrex] est un borosilicate de composition`` (C. DUVAL, Le Verre, 1966, p. 13); verre boro-silicaté (F. MEYER, P. GRIVET, Le Verre, Paris, P. U. F., 1947, p. 37); borohydrures d'aluminium, de radium, de potassium (Hist. gén. des sc., t. 3, vol. 2, 1964, p. 429); borotartiate d'aluminium (P. LEBEAU, G. COURTOIS, Traité de pharm. chim., t. 1, 1929, p. 761).
Prononc. :[ ]. Homon. bord, bort. Étymol. et Hist. 1809 (GAY-LUSSAC, THENARD, Extr. d'un mém. lu à l'Institut National le 14 nov. 1808 dans Mém. de phys. et de chim. de la Société d'Arcueil, t. 2, p. 315 : tout nous prouve que le radical boracique, que nous nous proposons d'appeler bore, est d'une nature particulière, et qu'on doit le placer à côté du charbon, du phosphore et du soufre). Tiré de borax par dér. régr., -ax étant senti comme suff. Le bore avait déjà été découvert en oct. 1807 par l'Anglais Sir Humphrey Davy, qui lui donna le nom de boracium (1808), puis de boron (1812) d'apr. la termin. de carbon (NED, s.v. boracium, boron). Fréq. abs. littér. :9.
]. Homon. bord, bort. Étymol. et Hist. 1809 (GAY-LUSSAC, THENARD, Extr. d'un mém. lu à l'Institut National le 14 nov. 1808 dans Mém. de phys. et de chim. de la Société d'Arcueil, t. 2, p. 315 : tout nous prouve que le radical boracique, que nous nous proposons d'appeler bore, est d'une nature particulière, et qu'on doit le placer à côté du charbon, du phosphore et du soufre). Tiré de borax par dér. régr., -ax étant senti comme suff. Le bore avait déjà été découvert en oct. 1807 par l'Anglais Sir Humphrey Davy, qui lui donna le nom de boracium (1808), puis de boron (1812) d'apr. la termin. de carbon (NED, s.v. boracium, boron). Fréq. abs. littér. :9.
]. Homon. bord, bort. Étymol. et Hist. 1809 (GAY-LUSSAC, THENARD, Extr. d'un mém. lu à l'Institut National le 14 nov. 1808 dans Mém. de phys. et de chim. de la Société d'Arcueil, t. 2, p. 315 : tout nous prouve que le radical boracique, que nous nous proposons d'appeler bore, est d'une nature particulière, et qu'on doit le placer à côté du charbon, du phosphore et du soufre). Tiré de borax par dér. régr., -ax étant senti comme suff. Le bore avait déjà été découvert en oct. 1807 par l'Anglais Sir Humphrey Davy, qui lui donna le nom de boracium (1808), puis de boron (1812) d'apr. la termin. de carbon (NED, s.v. boracium, boron). Fréq. abs. littér. :9.DÉR. Borure, subst. masc. ,,Combinaison du bore avec un corps simple`` (Lar. encyclop.). Borure de magnésium. — Seule transcr. dans LITTRÉ et DG : bò-rùr. — 1re attest. 1820 (NYSTEN Suppl.); dér. de bore, suff. -ure.
BBG. — PAMART (P.). De l'alchim. à la chim. Vie Lang. 1969, p. 138.
bore [bɔʀ] n. m.
ÉTYM. 1809, Gay-Lussac et Thénard; découvert en 1807 par Sir Humphrey Davy, qui l'avait nommé boracium (1808), puis boron (1812) d'après carbon « carbone »; de borax.
❖
♦ Chim. Corps simple métalloïde, symb. B no at. 5, température de fusion 2 510 °C, densité 2,34 ou 2,37 (variétés cristalline et amorphe). || Bore cristallisé pur. || Le bore est transparent à certains rayonnements infrarouges. || Dérivés du bore. ⇒ Borique (acide), borax. || Intoxication due au bore. ⇒ Borisme. || Certains composés du bore sont utilisés dans la fabrication de verres spéciaux (pyrex) et dans l'industrie nucléaire.
❖
DÉR. Borique, borisme, borure.
HOM. Bord, bort.
Encyclopédie Universelle. 2012.