GLUCIDES
Les glucides, les lipides et les protides sont les trois grandes familles de composés naturels dont le rôle biologique est fondamental. Glucides est le nom moderne de ce que l’on appelait autrefois «saccharides». En fait, les glucides comprennent, outre des matières naturelles sucrées, dont le type est la saccharose, un certain nombre d’analogues purement synthétiques et de nombreux dérivés, naturels pour la plupart. On les divise en trois classes: oses, holosides, hétérosides. Seuls les oses ne sont pas hydrolysables. L’hydrolyse des holosides fournit exclusivement des oses ou des mélanges d’oses (fig. 1), alors que celle des hétérosides donne, à côté d’un ou plusieurs oses, une ou plusieurs molécules d’aglycones qui ne sont plus des oses.
Bien que la structure des glucides soit souvent complexe, leur chimie ne fait intervenir que deux fonctions fondamentales: la fonction carbonyle (aldéhyde ou cétone) et la fonction alcool.
Les oses se divisent en «oses normaux» dont la formule «linéaire» est CH2OH 漣(CHOH)n -2 漣CHO pour les aldoses (n 礪 3) ou CH2OH 漣 (CHOH)n -3 漣CO 漣CH2OH pour les cétoses (avec n 礪 4). Les «oses anormaux» sont également nombreux. Ils diffèrent des premiers par l’absence d’une ou de plusieurs fonctions alcool remplacées par des fonctions hydrocarbure (désoxyoses), la place différente du groupe cétonique dans le cas des cétoses, la ramification éventuelle de la chaîne qui fait apparaître deux fonctions alcool primaire et une fonction alcool tertiaire, l’oxydation plus profonde (osones), qui crée plusieurs fonctions carbonyles, l’impossibilité d’une cyclisation (oses trop pauvres en carbone).
Les oses normaux ont pour formule brute (CH2O)n ; les holosides qui en dérivent s’écrivent Cn (H2O)p (p 麗n ). Cette remarque les avait fait désigner autrefois par «hydrates de carbone», nom impropre; il exclut en effet les désoxyoses qui gardent un grand nombre des propriétés des oses normaux, et s’applique à des composés qui n’ont plus que de lointains rapports avec les oses.
Au sein du métabolisme des glucides, le D-glucose joue un rôle fondamental dans le fonctionnement des êtres vivants: c’est un générateur d’énergie facilement utilisable (dans un organisme animal, un gramme de glucides produit 17,1 kilojoules, cependant qu’un gramme de lipides, de densité plus faible, en fournit 37,6). Cette énergie, transitoirement conservée sous forme de substances « riches en énergie», dont le principal représentant est l’adénosine-triphosphate (ATP), peut être restituée par couplage de réactions d’hydrolyse de l’ATP en ADP (adénosine-diphosphate) ou AMP (adénosine-monophosphate) avec des réactions qui consomment de l’énergie [cf. BIOÉNERGÉTIQUE]. D’autre part, le métabolisme du glucose fournit un matériel de base pour la synthèse de substances indispensables à l’activité cellulaire: ribose (acides nucléiques), ribulose-1,5-diphosphate (assimilation chlorophyllienne), inositol (phospholipides), acide ascorbique (vitamine C de certains animaux), acides aminés (alanine, acides aspartique et glutamique), noyau benzénique (phénylalanine, tyrosine, tryptophane) et éventuellement acides gras (lipides) et stérols (membranes cellulaires, hormones...).
1. Oses
On peut les définir à partir des hexoses C6H126 dont le plus connu est le D-glucose.
Glucose
Le glucose constitue des efflorescences à la surface des fruits secs (raisin, figue). C’est le principal constituant du miel. Il se forme dans l’hydrolyse de nombreux holosides, polyosides, hétérosides.
L’industrie le prépare par hydrolyse acide de l’amidon. L’hydrolysat, neutralisé, est concentré sous vide; on obtient le «glucose massé». Celui-ci est purifié, après redissolution, passage au noir animal, et recristallisation. On obtient le glucose pur du commerce, constitué de cristaux blancs, mais qui est généralement un mélange de trois formes: le glucose- 見 hydraté, obtenu pur par une recristallisation très lente de solutions saturées; le glucose 見-anhydre résultant de l’élimination sous vide de l’eau d’hydratation du précédent; le glucose- 廓 qui se dépose lors d’addition d’éthanol aux solutions aqueuses saturées.
Les solutions fraîches de glucose- 見 ont un pouvoir rotatoire de + 1120; celles de glucose- 廓, de + 190; mais, après 24 heures environ, les deux solutions ont un même pouvoir rotatoire de + 520. Il existe donc, en solution, un équilibre entre les glucoses 見 et 廓. Ce phénomène porte le nom de «mutarotation». Si on admet que les glucoses 見 et 廓 sont les seuls constituants de la solution, on voit que le glucose- 廓 est, en gros, deux fois plus abondant que le glucose- 見 à l’équilibre.
Les glucoses ( 見-anhydre et 廓) ont des points de fusion différents, voisins de 155 0C. Le glucose hydraté fond un peu au-dessus de 100 0C, sans point de fusion défini.
Établissement de la formule chimique
L’analyse et la cryoscopie assignent au glucose la formule brute C6H126. Il est transformé en hexane normal, par chauffage en tube scellé avec de l’eau, de l’acide iodhydrique et du phosphore rouge (réduction universelle de Marcelin Berthelot), ce qui montre que les 6 carbones sont disposés linéairement.
L’hydrogénation catalytique transforme les glucoses 見 ou 廓 en un hexa-alcool, le sorbitol (CH2OH 漣(CHOH)4 漣CH2OH); cela fait pressentir que les glucoses sont des aldéhydes ou des cétones provenant, théoriquement, de la déshydrogénation partielle du sorbitol, soit:

Or, les glucoses 見 ou 廓 s’oxydent (Br2 + H2O) en un acide, l’acide gluconique, à 6 atomes de carbone; d’ailleurs l’oxydation plus poussée (HNO3) conduit à un diacide, l’acide saccharique ayant, lui aussi, 6 atomes de carbone.
Ces constatations tendent à démontrer l’existence d’une fonction aldéhyde (tabl. 1, formule 1), qui semble également attestée par la formation, sous l’influence de l’hydroxylamine, d’une oxime déshydratable en nitrile (réaction qui permet de distinguer les aldéhydes des cétones).
Mais d’autres arguments sont beaucoup moins favorables. Traités par ICH3 en milieu alcalin, les glucoses 見 et 廓 sont transformés en dérivés pentaméthylés qui devraient être:

Or, d’une part les glucoses 見 et 廓 conduisent à des dérivés pentaméthylés différents et non en équilibre entre eux; d’autre part ces dérivés pentaméthylés ne sont pas réducteurs et ne présentent aucune des propriétés des aldéhydes.
De plus, on connaissait des diastéréo-isomères (isomères optiquement actifs ne différant que par les positions des substituants d’un carbone asymétrique) naturels du glucose (mannose, galactose); à la formule (1), qui comprend 4 carbones asymétriques, correspondent 16 diastéréo-isomères [cf. STÉRÉOCHIMIE]. La première hypothèse était de considérer les glucoses 見 et 廓 comme deux de ces diastéréo-isomères. Mais si l’on considère comme tels les isomères 見 et 廓 des autres aldohexoses à partir du moment où des diastéréo-isomères artificiels ont pu être préparés, le nombre des variétés était bien supérieur à 16 et peu inférieur à 32. Il fallait donc envisager un cinquième carbone asymétrique. Les formes 見 et 廓 étant en équilibre chez les glucoses 見 et 廓, et non chez leurs dérivés pentaméthylés, il était normal de penser que le cinquième carbone asymétrique avait une constitution labile chez les glucoses, et stable chez leurs dérivés pentaméthylés. Cela est réalisé dans l’hypothèse d’une cyclisation.
En effet, la fonction hémiacétal est peu stable et en équilibre avec la forme linéaire mais, dans la cyclisation, le carbone hémiacétalique peut prendre deux configurations conduisant aux isomères 見 et 廓.
On pourrait concevoir une autre cyclisation conduisant à un cycle pentagonal (furannose). En ce qui concerne le glucose, cette forme ne semble pas intervenir.
Ces hypothèses ont été complètement confirmées par l’étude physico-chimique des glucoses 見 et 廓.
Les formules des deux hexoses méthylés se déduisent alors de la formule (1) par substitution de groupements 漣OCH3 aux groupements OH.
La fonction hémiacétal étant bloquée sous forme d’acétal, fonction stable, on conçoit à la fois que les dérivés pentaméthylés ne sont pas réducteurs et que leurs formes 見 et 廓 ne subissent plus la mutarotation.
En ce qui concerne les glucoses 見 et 廓, le passage réversible en solution aqueuse par la forme aldéhydique explique cette mutarotation. Toutefois, à l’équilibre, la concentration de cette forme aldéhydique est si faible que les critères physiques (ultra-violet, Raman) ne permettent pas de la déceler; mais les réactifs de la fonction aldéhyde, grâce à un déplacement de l’équilibre, agissent comme si tout le glucose était sous cette forme. Les formes solides 見 et 廓 sont, évidemment, exclusivement pyrannosiques.
Configuration stérique
Le problème est double: il s’agit d’établir les configurations stériques des 4 atomes de carbone asymétrique stables, puis celles du cinquième carbone asymétrique par lequel diffèrent les glucoses 見 et 廓.
Dans le premier cas, il est plus simple de raisonner sur les formules linéaires: CH2OH 漣(CHOH)4 漣CHO.
On utilise trois transformations:
– sous l’influence de l’hydroxylamine, un aldohexose conduit à une oxime déshydratable en nitrile:
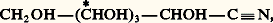
lequel, sous l’action de l’oxyde d’argent, se décompose en HCN et l’ose inférieur:
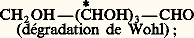
– un aldohexose fixe HCN pour engendrer deux cyanhydrines:

se distinguant par la configuration du carbone marqué d’une croix; ces nitriles sont hydrolysés en acides:

Les deux isomères sont séparés et réduits par l’amalgame de sodium en deux aldoheptoses isomères:

Ces trois transformations ne modifient pas la configuration des carbones asymétriques groupés, mais la deuxième supprime le carbone asymétrique voisin du groupe CHO.
Le glucose aldéhydique sera provisoirement écrit:

les quatre points représentant les carbones asymétriques dont la configuration sera progressivement établie.
Trois dégradations de Wohl transforment le glucose en aldéhyde glycérique, CH2OH 漣CHOH 漣CHO, dont il n’existe que deux inverses optiques. Celui provenant de la dégradation du glucose est dextrogyre; on l’appelle, par définition, aldéhyde D-glycérique et on l’a d’abord écrit arbitrairement CH2OH 漣 face="EU Caron" ゲCHO [cf. STÉRÉOCHIMIE].
Cette constitution s’est révélée exacte lorsqu’on a réussi à établir sa configuration absolue. Dès lors, tous les aldoses dont la dégradation jusqu’au stade C3 conduit à l’aldéhyde D-glycérique sont dits appartenir à la série D; désormais le glucose sera appelé D-glucose; il est dextrogyre sous ses formes 見 et 廓, mais ce n’est qu’une heureuse coïncidence qui ne se répète que fortuitement pour les autres aldoses de la série D.
Dégradé une fois de moins, le D-glucose conduit à un aldotétrose, le D-érythrose que l’oxydation transforme en acide mésotartrique, inactif, écrit:

La configuration des deux premiers carbones asymétriques est donc:
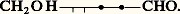
Une seule dégradation du D-glucose le transforme en D-arabinose, oxydable en un acide optiquement actif qui ne peut être que:
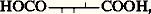
d’où, pour le D-glucose, la structure partielle:

Mais le fait que l’acide D-saccharique est optiquement actif ne nous renseigne pas sur la structure du quatrième carbone asymétrique; en effet, les deux acides:

sont tous les deux optiquement actifs.
Deux méthodes permettent de résoudre le problème:
– On passe aux deux aldoheptoses supérieurs (Kiliani-Fischer); oxydés, ils conduisent à deux acides, dont un seul est optiquement actif:

Cela n’est compatible que si le quatrième carbone asymétrique a la configuration inverse de celle de son voisin de gauche; cela impose au D-glucose la projection:
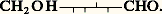
– On peut lever autrement l’ambiguïté sur le quatrième carbone asymétrique: dans les formules ci-dessus, à (1) correspond un seul aldohexose, alors qu’à (2) en correspondent deux: le D-glucose et son diastéréo-isomère:

Ce dernier a pu être préparé; c’est le L-gulose, ce qui assigne à l’acide saccharique la formule (2), et au D-glucose, la formule proposée.
La configuration du cinquième carbone asymétrique qui distingue les glucoses 見 et 廓 est plus délicate à déterminer. Il a été établi que les structures respectives sont celles des formules (2) du tableau 1.
Le cycle hexagonal est sous forme «chaise»; les hydroxyles, le groupe CH2OH occupent, au maximum, les positions équatoriales [cf. STÉRÉOCHIMIE].
Autres aldohexoses
La configuration stérique des diastéréo-isomères du D-glucose peut être établie de la même façon; c’est beaucoup plus simple pour le galactose provenant de l’hydrolyse du lactose (ou sucre de lait). Trois dégradations lui assignent la série D; le diacide provenant de l’oxydation (acide mucique) est inactif; ce ne peut être que:

Or, en oxydant l’aldopentose de première dégradation (D-lyxose), on obtient un acide optiquement actif, ce qui ne laisse possible, pour le D-galactose, que la configuration:
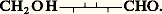
Le travail est abrégé par les remarques fondamentales suivantes:
– si deux aldohexoses sont inverses optiques, l’un appartient à la série D, l’autre à la série L; l’inverse optique du D-glucose, le L-glucose, s’écrit:

– si deux aldohexoses différents s’oxydent en un même diacide, ils diffèrent par la permutation de CH2OH et de CHO; en conséquence, le L-gulose conduisant, comme le D-glucose, à l’acide saccharique, est:

– on appelle aldohexoses épimères deux diastéréo-isomères se différenciant par la configuration du carbone asymétrique voisin de CHO. Or, la phénylhydrazine 淋-NH-NH2 transforme les composés R 漣CHOH 漣CHO en osazone R 漣C (= N 漣 NH 漣 淋) 漣 CH = N 漣 NH 淋, faisant disparaître l’asymétrie du carbone épimère; en conséquence, deux sucres épimères conduisent à la même osazone. L’aldohexose dont l’osazone est identique à celle du D-glucose s’écrit:

C’est le D-mannose.
De proche en proche, on arrive ainsi à établir la structure de l’un des inverses optiques des autres aldohexoses; cette méthode suppose que tous les diastéréo-isomères soient à la disposition du chimiste: or, à peu près seuls, le D-glucose, le D-galactose et le D-mannose sont facilement accessibles à partir des produits naturels. Il a été nécessaire de préparer les autres par des synthèses partielles.
Épimérisation
L’épimérisation directe (passage du D-glucose au D-mannose) peut se faire en milieu alcalin mais, à l’équilibre, ce sont les cétohexoses qui dominent. Par contre, l’acide D-gluconique:

est rapidement mis en équilibre par les alcalis avec l’acide D-mannonique:

Ces acides sont séparés; le premier par réduction redonne le D-glucose, et le second conduit au D-mannose; la méthode est applicable aux autres aldohexoses épimères.
Permutation entre CH2OH et CHO
L’aldohexose est oxydé en diacide et celui-ci est soumis à une réduction incomplète; on aboutit en principe à un mélange de l’aldose de départ et de l’ose permuté; c’est ainsi que la réduction de l’acide saccharique fournit, à côté de très peu de D-glucose, une forte majorité de L-gulose.
La constitution des aldohexoses naturels étant:

ces méthodes ne permettent pas l’accès aux aldohexoses:
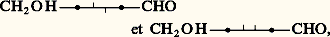
car les deuxième et troisième carbones asymétriques ne sont pas modifiés.
Toutefois on peut passer du D-galactose au galactose racémique:

en oxydant le D-galactose, et en réduisant l’acide mucique (inactif) ainsi obtenu; la fermentation alcoolique détruit le D-galactose, d’où un accès au L-galactose.
D’autre part, l’hydrolyse de la gomme arabique fournit un aldopentose, le L-arabinose:

La synthèse de Kiliani-Fischer transforme celui-ci en un mélange de L-glucose et de L-mannose.
C’est grâce à ces travaux laborieux qu’Emil Fischer a réussi, il y a de cela une centaine d’années, à préparer tous les aldohexoses à partir du D-glucose, du D-galactose et du L-arabinose.
Cétohexoses
Les cétoses normaux ont pour formule plane:
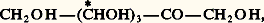
qui fait prévoir huit diastéréo-isomères, inverses optiques deux à deux.
Chacun existe sous deux formes pyranniques 見 et 廓 (tabl. 1, formule 2). Leur constitution se déduit de celle des aldohexoses.
L’hydrogénation conduit à deux hexitols épimères car la réduction du carbonyle cétonique n’est pas stéréospécifique.
La phénylhydrazine les transforme en osazones:

Il s’agit de l’osazone déjà commune à deux aldohexoses épimères. Cette transformation ne touchant pas la configuration des carbones asymétriques groupés, on conclut, par exemple, que le cétohexose dont l’osazone est identique à celle du D-glucose ou du D-mannose est:
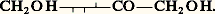
C’est le lévulose ou D-fructose, ose fortement lévogyre bien qu’il appartienne à la série D. Présent dans le miel, formé partiellement dans l’hydrolyse du saccharose, il prend naissance, à peu près seul, dans l’hydrolyse de l’inuline qui en est un polyoside.
Un autre cétohexose important est le L-sorbose:

Il se forme dans les sorbes à maturité par l’oxydation biochimique (bactérie du sorbose) du D-sorbitol, généralement préparé par hydrogénation catalytique du D-glucose. Cette oxydation biochimique peut être réalisée in vitro , mais elle exige que le polyol puisse être écrit:
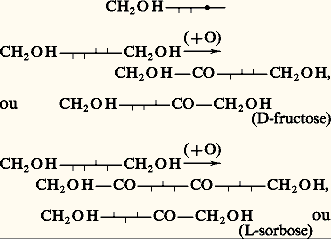
Les cétohexoses réduisent la liqueur de Fehling et le nitrate d’argent ammoniacal; tous les oses sont réducteurs. Ils sont tous résinifiés par les alcalis, ce qui les distingue de certains holosides. La plupart des hexoses naturels subissent, entre autres, la fermentation alcoolique; leurs inverses optiques, et la plupart des hexoses synthétiques, ne subissent pas ces fermentations.
Autres oses
Quelques aldoheptoses ont été rencontrés à l’état naturel chez les champignons supérieurs; d’autres, ainsi que les aldo-octoses, -nonoses, -décoses, résultent de synthèses de Kiliani-Fischer.
Les aldo et céto-pentoses sont tous connus; les uns sont naturels; les autres proviennent d’épimérisations de ces derniers ou de dégradations des aldohexoses. Il en est de même pour les aldo et céto-tétroses.
Parmi les oses aberrants, nous citerons seulement la série complète des seize méthylpentoses ( 見 et 廓):

comprenant des produits naturels et des produits synthétiques, et le désoxy-2 D-ribose:

qui joue un rôle biologique considérable.
2. Holosides
Diholosides
On désigne ainsi des glucides dérivant, théoriquement, de l’élimination d’une molécule d’eau entre deux oses sous forme cyclique. En nous limitant au cas où ces deux oses sont des aldohexoses, on prévoit un très grand nombre d’isomères, cent dans le cas le plus général. Toutefois, tous les diholosides naturels et la très grande majorité des diholosides synthétiques utilisent l’un au moins des hydroxyles hémiacétaliques (1) dans cette déshydratation.
La représentation des diholosides utilise le symbole 麗礪; de part et d’autre figurent les noms des oses générateurs, et, à l’intérieur, les numéros des hydroxyles utilisés dans l’anhydrisation. Le corps représenté par la formule 4 est le D-galactopyrannose- 廓 麗14 礪D-glucopyrannose- 見.
Il existe deux types fondamentaux de diholosides: les diholosides 麗1-1 礪 appelés aussi osido-osides (ou diholosides non réducteurs) et les diholosides 麗1-n 礪 ou 麗n -1 礪 (n = 2, 3, 4 ou 6) appelés aussi osido-oses ou diholosides réducteurs.
En effet, les premiers ne renferment que des fonctions alcool et une fonction double acétal, ne s’ouvrant pas en milieu alcalin. Ils ne réduisent pas le nitrate d’argent ammoniacal et la liqueur de Fehling, alors que les seconds possèdent une fonction hémiacétal libre, donc réductrice.
La structure des diholosides s’établit ainsi: l’hydrolyse fournit deux oses; dans le cas des diholosides non-réducteurs, une seule ambiguïté subsiste en ce qui concerne la structure des carbones intéressés par la déshydratation: 見見 , 見廓 , 廓見 ou 廓廓 . L’additivité approchée des pouvoirs rotatoires permet de lever cette ambiguïté.
En ce qui concerne les diholosides réducteurs, on méthyle tous les hydroxyles libres, puis on hydrolyse le dérivé octométhylé. L’ose engagé en 麗 1 est libéré sous forme de tétra-méthyl-ose; celui engagé en n 礪 est libéré sous forme de tri-méthyl-ose; l’étude de ce dernier fixe facilement la valeur de n .
Tous les diholosides sont hydrolysés par ébullition avec les acides dilués; mais, dès la température ordinaire, l’hydrolyse peut se faire sous l’influence d’enzymes spécifiques; c’est ainsi, par exemple, que les 見-glucosidases hydrolysent tous les diholosides D-glucopyrannose- 見 麗 1-n 礪... et que les 廓-galactosidases hydrolysent tous les diholosides D-galactopyrannose- 廓 麗 1-n 礪...
Si l’on engage, sous forme de diholoside, un cétohexose, le numérotage est un peu différent; dans les cétoses, le carbone hémiacétalique est numéroté 2; c’est ainsi que le plus connu des diholosides (le saccharose ou sucre de canne ou de betterave) est le D-glucopyrannose- 見 麗 1-2 礪 D-fructofurannose- 廓.
Cet holoside est non réducteur, mais le devient, après hydrolyse acide, car il est transformé en D-glucose et D-fructose, tous deux réducteurs. Cette hydrolyse fait passer le pouvoir rotatoire de + 660 à 漣 200; en effet, le D-glucose à l’équilibre (+ 520) est bien moins dextrogyre que le D-fructose à l’équilibre (face=F0019 漣 720) n’est lévogyre. C’est la raison qui a fait donner le nom d’inversion à l’hydrolyse de saccharose. Ce terme est parfois conservé dans les cas où le pouvoir rotatoire change de valeur sans changer de signe.
La plupart des diholosides sont nettement sucrés. Les diholosides non-réducteurs ne sont pas résinifiés par les alcalis, alors que les diholosides réducteurs se comportent vis-à-vis d’eux comme les oses. Seuls les diholosides réducteurs (n 2) fournissent, avec la phénylhydrazine, des osazones.
Polyholosides
Les polyholosides constituent un vaste groupe de produits naturels très répandus chez les êtres vivants. Sous l’angle fonctionnel, ces substances jouent le rôle de réserves nutritives ou de matériaux structuraux et protecteurs.
Leurs facultés protectrices se manifestent dans les exosquelettes des Crustacés et des Insectes, les parois et les capsules de certaines bactéries et enfin les gommes et exsudats qui oblitèrent les blessures chez beaucoup de plantes. Comme matériau structural, ils contribuent à maintenir la rigidité des plantes et forment les constituants principaux de leurs mucilages, tandis que, chez les animaux, ils entrent dans la composition des cartilages et des liquides intra-articulaires.
Les facteurs responsables de la spécificité des groupes sanguins, certains facteurs antigéniques et les agents anticoagulants des tissus animaux sont également des polyholosides.
Du point de vue chimique, les polyholosides sont des produits de condensation d’oses et de dérivés d’oses unis par des liaisons glycosidiques. On distingue: d’une part, les polyholosides homogènes (homopolyholosides ou homoglucanes), constitués par un seul type ose, et très largement distribués dans la nature (cas de l’amidon, du glycogène et de la cellulose); d’autre part, les polyholosides hétérogènes (hétéropolyholosides ou hétéroglucanes), qui peuvent résulter de l’association de deux oses ou de dérivés d’oses distincts.
Leur structure moléculaire peut être soit linéaire comme dans la cellulose, soit ramifiée comme dans la gomme arabique, soit encore de type mixte comme dans l’amidon.
Polyholosides homogènes. Polymères du glucose (glucanes)
Les polymères du glucose sont la cellulose, l’amidon, le glycogène et les dextranes.
Cellulose
La cellulose pure est représentée par les fibres de coton qui sont des cellules dont la longueur varie entre 10 et 50 mm (cf. fig. 3 de l’article FIBRES TEXTILES). Les autres sources de cellulose sont le bois, le lin, le chanvre et la paille. Les usages industriels de la cellulose se fondent sur sa nature fibreuse, qui dépend elle-même de la structure moléculaire.
La cellulose est le constituant principal de la paroi cellulaire des plantes et, comme telle, la substance naturelle la plus abondante dans la biosphère. Sa fonction biologique se rattache à ses propriétés: insolubilité, inertie chimique, rigidité. L’analyse chimique (méthylation, hydrolyse acide) a permis de montrer que la cellulose est composée de longues molécules dans lesquelles des résidus D-glucosyl sont reliés par des liaisons 廓 (14), l’unité élémentaire étant le cellobiose (tabl. 2, formule 1).
La longueur de la chaîne moléculaire, exprimée par le nombre moyen d’unités glucosyl, représente le degré de polymérisation (D.P.); elle peut être évaluée par des méthodes physiques et chimiques, parmi lesquelles la mesure de la viscosité. Les résultats dépendent de l’origine de la cellulose, du traitement subi par ce matériau et de la méthode de mesure. Le D.P. de la cellulose naturelle est considéré comme supérieur à 3 000. La longueur de la chaîne semble varier d’une plante à une autre, bien que des différences puissent survenir du fait de ruptures au cours de l’isolement. Les nitrocelluloses sont employées comme explosifs et comme matières plastiques.
Amidon
De grandes quantités d’amidon sont mises en réserve par les organismes végétaux. Au microscope, l’amidon apparaît sous forme de grains. Les amidons constituent un aliment pour la plante; ils entretiennent en effet la vie de la tige ou du tubercule pendant le repos hivernal et assurent le développement de l’embryon au cours de la germination [cf. GRAINE]. Les principales sources commerciales de l’amidon sont le maïs, la pomme de terre et le manioc. La plupart des amidons sont formés d’un mélange de deux constituants: l’amylose, dont la structure est linéaire, et l’amylopectine, dont la structure est ramifiée (tabl. 2, formules 2 et 3). La proportion de ces fractions est caractéristique des espèces. Ainsi, les amidons du blé et de la pomme de terre renferment 15 à 30 p. 100 d’amylose, tandis que les amidons de certaines variétés de maïs, de riz et de sorgho, en Chine, sont presque entièrement constitués d’amylopectine. En revanche, les amidons de certains pois et d’autres variétés de maïs renferment surtout de l’amylose.
Contrairement à la cellulose, les amidons peuvent être dispersés dans l’eau chaude. Mis en suspension dans l’eau et chauffés à une température critique, les grains d’amidon gonflent et forment un empois qui devient une pâte en refroidissant. L’amylose serait responsable de la gélification, alors que l’amylopectine contribuerait à donner à la pâte sa cohésion.
L’amidon est hydrolysé par les acides dilués et par des enzymes (amylases). L’hydrolyse acide totale fournit du D-glucose, tandis que l’hydrolyse enzymatique fournit surtout un diholoside, le maltose. Des fragments de poids moléculaire plus élevé, les dextrines, sont obtenus par hydrolyse acide contrôlée et action de la température.
L’amylose (tabl. 2, formule 2) est formée d’une longue chaîne d’unités D-glucosyl (environ 200 à 300) assemblées par des liaisons 見 (14). Une telle association moléculaire, dans laquelle les groupements 漣CH2OH sont placés du même côté, ne peut favoriser la structure linéaire de l’amylose, celle-ci a donc tendance à s’enrouler en hélice.
Dans l’amylopectine, les unités glucosyl sont assemblées de manière à réaliser une structure très ramifiée (tabl. 2, formule 3). Aux embranchements, la molécule de D-glucose s’attache aux autres unités, en position 6, par une liaison 見 (16), ou encore en position 1 et 4: 見 (14). En plus de ces connexions principales existent, comme on l’a découvert ultérieurement, un petit nombre de liaisons 見 (13).
Les molécules ramifiées ou non ramifiées qui constituent les amidons se distinguent par la couleur des complexes apparaissant en présence d’iode qui colore l’amylose en bleu et l’amylopectine en violet. Elles peuvent être séparées par l’alcool amylique et par de nombreux composés organiques simples qui précipitent l’amylose sous forme de complexes microcristallins. La connaissance de la biogenèse de l’amidon a été améliorée par la synthèse au laboratoire de l’amylose et de l’amylopectine. On est parvenu à ce résultat en utilisant deux enzymes extraites de divers produits naturels, tels que la pomme de terre. Il semblerait que la polymérisation du D-glucose, qui donnera l’amidon des plantes et le glycogène des animaux (ainsi que la combinaison de D-glucose avec le D-fructose pour former du saccharose), soit effectuée par l’intermédiaire d’un ester phosphorique du D-glucose: l’ 見 D-glucopyrannose 1-phosphate.
Glycogène
Très voisins des amylopectines, les glycogènes constituent les réserves glucidiques des tissus animaux. Ils sont aussi produits par les levures, les champignons et diverses algues. Comme les amylopectines, leurs poids moléculaires sont très élevés et leurs structures moléculaires sont ramifiées et formées d’unités de D-glucose. Cependant, les chaînes qui les composent sont plus nombreuses et plus courtes que celles des amylopectines (tabl. 2, formule 3). Par suite, leurs molécules sont plus compactes, plus arborescentes, plus solubles et leurs solutions moins visqueuses.
Dextranes
Les dextranes se trouvent dans les matières gluantes produites par la croissance de certaines bactéries, tel Leuconostoc mesenteroides , sur milieux saccharosés. Ils ont un poids moléculaire élevé et se composent d’unités D-glucosyl unies principalement par des liaisons 見 (16). Des dextranes, partiellement hydrolysés et ajoutés à une solution saline physiologique, sont employés comme succédanés du plasma sanguin dans les traitements et l’état de choc.
Polyholosides homogènes. Polymères d’autres sucres
Polymères d’autres aldoses
Les galactanes, les mannanes, les xylanes (tabl. 2, formule 4) et les arabanes sont des polyholosides qui existent principalement dans les parois cellulaires des plantes. Ce sont les principaux constituants des hémicelluloses et des matières pectiques.
Polymères du fructose (fructanes)
Les fructanes constituent, tout comme l’amidon, la réserve glucidique de certaines plantes, particulièrement Composées et Graminées. L’inuline , très répandue dans de nombreux organes de plantes (artichaut, dahlia), se range dans ce groupe. Elle est composée de 30 à 40 résidus D-fructofuranosyl, unis par des liaisons 廓 (12), et de 1 à 2 unités D-glucopyranosyl placées en bout de chaîne (tabl. 2, formule 5).
D’autres variétés de fructanes existent dans les feuilles d’herbages et sont appelées levanes . Leurs molécules sont des chaînes linéaires d’environ vingt résidus fructosyl unis par des liaisons 廓 (26) et terminés par un résidu saccharose. Des levanes du même genre sont produits par des microorganismes, comme Bacillus subtilis .
Polymères d’osamines: la chitine
La chitine est un polyholoside des exosquelettes des Crustacés (25 p. 100 de chitine et 75 p. 100 de carbonate de calcium dans la carapace d’un crabe) et des Insectes. On la trouve aussi chez certains Champignons. La structure de la chitine rappelle celle de la cellulose; elle consiste en de longues chaînes de résidus de N-acétylglucosamine avec liaisons 廓 (14) [tabl. 2, formule 6]. La chitine est une substance inerte, difficile à hydrolyser.
Polyholosides hétérogènes
Hémicelluloses
Les hémicelluloses sont un vaste groupe de polyholosides, insolubles dans l’eau, associés à la cellulose dans les parois cellulaires des plantes. Leur nature et leur composition ne sont pas complètement établies. Elles renferment une grande quantité de xylanes, polyholosides formés d’unités D-xylopyranosyl fixées par des liaisons 廓 (14), ainsi que diverses combinaisons de L-arabinose, D-glucose, D-galactose, D-mannose, acide D-glucuronique et acide D-galacturonique. La solubilité des hémicelluloses dans les solutions alcalines diluées permet de les séparer de la cellulose.
Matières pectiques
Situées dans les parois cellulaires et dans les ciments intercellulaires (lamelles moyennes) des tissus végétaux, les matières pectiques ont un pouvoir gélifiant élevé. On les extrait de l’écorce de citron, de la pomme et de la betterave. Elles sont formées d’acide poly-D-galacturonique, partiellement estérifié par le méthanol, et associé à des arabanes et à des galactanes (tabl. 2, formule 7).
Gommes végétales
Les gommes constituent un groupe de polyholosides à structure très complexe donnant des solutions à viscosité élevée. La gomme adragante est un exsudat produit par les Astragalus (ordre des Légumineuses). La gomme arabique, provenant de certaines espèces du genre Acacia , contient du L-arabinofuranose, du D-galactopyranose, du L-rhamnose (6-désoxy L-mannose) et de l’acide D-glucuronique.
Mucopolysaccharides
On comprend sous cette dénomination un vaste groupe de substances très importantes en physiologie et qui englobe: les acides chondroïtine-sulfuriques du cartilage (tabl. 2, formule 8), l’héparine (tabl. 2, formule 10), substance anticoagulante localisée dans les mastzellen ou mastocytes du tissu conjonctif des animaux, l’acide mucoïtine-sulfurique de la muqueuse gastrique et de la cornée, l’acide hyaluronique (tabl. 2, formule 9) du tissu conjonctif, du liquide synovial, du cordon ombilical de divers tissus animaux et des capsules de certaines bactéries. Ces polysaccharides sont formés par la polymérisation d’une unité disaccharidique elle-même formée d’une osamine (D-glucosamine ou D-galactosamine) et d’un acide uronique (acide D-glucuronique le plus souvent). Les osamines portent des groupements sulfate ou acétamide. Les polysaccharides sont unis à des protéines par liaisons électrovalentielles.
Polyholosides bactériens
La spécificité antigénique de certaines bactéries est souvent liée à la possession d’une capsule plus ou moins épaisse et compacte, dont la nature est déterminée génétiquement. La purification des capsules a montré que ce sont des polyholosides de poids moléculaire élevé et de composition plus ou moins complexe. De nombreux antigènes capsulaires de pneumocoques contiennent, en dehors du glucose et du galactose, des pentoses et des sucres aminés.
La paroi bactérienne, rigide, perméable à l’eau, aux sels et aux métabolites, confère à la bactérie sa forme. Elle contient des antigènes spécifiques responsables de l’agglutination somatique type 0. Elle est indispensable à la fixation et à la pénétration des bactériophages dans la cellule. Elle est constituée par un muco-complexe composé de sucres, de lipides et d’acides aminés, dont la nature et les proportions relatives varient sensiblement suivant l’organisme, et en particulier selon qu’il s’agit de bactéries Gram-positives ou Gram-négatives. Les sucres – surtout rhamnose, arabinose, glucose, plus rarement galactose, mannose – forment 20 à 60 p. 100 du poids sec. On trouve aussi des sucres aminés: N-acétylgalactosamine et N-acétylglucosamine. La glucosamine entre dans la composition de l’acide muramique (éther-oxyde de l’acide lactique et de la N-acétylglucosamine), constituant important de la structure macromoléculaire de la paroi bactérienne. On a extrait du bacille diphtérique un polysaccharide constitué par deux molécules de D-galactose, une molécule de D-mannose et trois molécules de D-arabinose.
3. Hétérosides naturels
Le terme hétéroside désigne des substances variées qu’on trouve surtout chez les plantes, et résultant de la combinaison du groupe réducteur d’un ose avec une substance non glucidique, l’aglycone. Pour les hétérosides naturels, l’aglycone se combine par l’intermédiaire soit d’un hydroxyle alcoolique ou phénolique (O-hétérosides), soit d’un thiol (S-hétérosides), soit d’un groupe aminé (N-hétérosides).
Propriétés générales
Le rôle biologique des hétérosides n’est pas clairement établi. On les isole des plantes sous forme de substances solides, en général cristallisées; la plupart ont un goût amer. Leurs solutions ont un pouvoir rotatoire (lévogyre en général). Sous l’action des acides, ces corps se scindent par hydrolyse en une ou plusieurs molécules d’oses et en une fraction non glucidique. Cette hydrolyse peut être aussi obtenue sous l’action d’enzymes spécifiquement actives sur un hétéroside ou sur une classe d’hétérosides apparentés. Les aglycones ont des structures variées, mais ils contiennent toujours un groupe hydroxyle (OH) qui assure la combinaison avec le glucide. Sur le plan chimique, un hétéroside est un acétal mixte et présente toutes les propriétés de ce genre de combinaison. La plupart des hétérosides naturels ont reçu une dénomination dérivée du nom botanique de la plante dont on les extrait. Ainsi, l’arbutoside ou arbutine, substance amère, incolore et cristallisée, obtenue à partir des feuilles de l’arbuste à feuilles persistantes, Arbutus uva ursi , fut retrouvé dans d’autres plantes, telles que le poirier (feuilles, écorce, racines). L’arbutoside est hydrolysé par les acides ou par les enzymes, telle la 廓-D-glucosidase (émulsine des amandes amères) en D-glucose et hydroquinone, qui représente l’aglycone; le nom chimique de l’arbutoside est donc para -hydroxyphényl 廓-D-glucopyranoside (tabl. 3, formule 1). Les hétérosides sont habituellement assez solubles dans l’eau; ce caractère permet de conclure, au moins à titre d’hypothèse de travail, que ce sont des substances transportables par la sève, d’où les idées suivantes:
– des substances toxiques ou inutiles pourraient être véhiculées vers l’écorce, les téguments des fruits et des graines, où elles seraient inactivées, voire éliminées;
– ces substances pourraient être réutilisées;
– certaines substances décoratives ou attractives, telles que les pigments des fleurs pourraient être synthétisées dans les feuilles, puis être transportées vers les fleurs et les fruits.
La théorie selon laquelle la formation des hétérosides dans la plante est un mécanisme de détoxication repose sur la découverte du rôle de 2-chloroéthanol (ClCH2 漣CH2OH), qui lève la dormance des tubercules et peut être transformé par les tissus végétaux en 2-chloroéthyl 廓-D-glucopyranoside. Les structures des hétérosides végétaux varient, d’une part, selon le sucre qu’ils renferment, d’autre part, selon la nature de l’aglycone. Leur présence dans les plantes explique leurs nombreux emplois (médicaments, poisons, colorants, condiments). Une fois leur structure chimique établie, beaucoup d’hétérosides – notamment les pigments, ainsi que des condiments et des médicaments d’origine végétale [cf. MÉDICAMENTS] – ont été obtenus par synthèse.
Principales classes
Hétérosides de phénols
La salicine est le principe actif de l’écorce de saule, qui fut longtemps utilisé contre la fièvre et les arthrites. Son hydrolyse libère le D-glucose et le saligénol (alcool orthohydroxybenzylique).
La phloridzine de l’écorce des Rosacées (pommier, poirier, cerisier, prunier) est le D-glucoside d’un aglycone phénolique complexe. Son administration au chien produit un diabète de type particulier, d’où l’intérêt de la phloridzine en médecine expérimentale.
Hétérosides d’alcools
Les hétérosides d’alcools aliphatiques sont représentés par des purgatifs, tels que la convolvuline et la jalapine ; les oses sont ici combinés avec les groupements hydroxyles d’acides gras hydroxylés.
Le 廓-D-glucopyranoside de l’alcool terpénique, nommé géraniol , se rencontre chez le Pelargonium odoratum .
Le stévioside , au goût remarquablement doux (contrairement à beaucoup d’hétérosides), est extrait des feuilles d’une Composée du Paraguay, Stevia rebaudiana . Il renferme trois unités D-glucose combinées l’une à l’autre; son aglycone (le stéviol) est un diterpénoïde.
Les saponines sont les hétérosides d’alcools aliphatiques très répandus chez les végétaux. Ce sont des produits moussants, toxiques pour le poisson mais non pour l’homme qui s’en nourrit, ce qui explique leur usage, chez certains peuples primitifs, dans la pêche. L’hydrolyse des saponines libère des oses variés, et parfois l’acide D-glucuronique. Les aglycones sont des dérivés des triterpènes ou des stérols (tabl. 3, formule 2).
D’autres hétérosides stéroïdiques d’intérêt considérable sont les glucosides cardiotoniques (cf. appareil CIRCULATOIRE). Puissants poisons pour le cœur à doses fortes, ils sont stimulants à petites doses. Les tribus africaines primitives les extraient des graines de Strophantus , du bois et de l’écorce des ouabaïas, pour enduire leurs flèches de ce poison. Les mieux connus en médecine moderne sont les hétérosides extraits des feuilles de Digitalis (tabl. 3, formule 3). D’autres sont obtenus à partir des fleurs de certains lys. L’hydrolyse des glucosides cardiotoniques libère une grande variété de sucres rares, mais leur action physiologique dépend de l’aglycone dont la structure s’apparente à celle des stéroïdes.
Les hétérosides cyanogénétiques se trouvent dans les amandes des pêches, des cerises, des prunes et dans d’autres organes de diverses plantes. Ce sont principalement des hétérosides du nitrile phénylglycolique (C6H5 漣 CHOH-CN). Hydrolysés par des enzymes spécifiques ou par des acides, ils libèrent: l’aldéhyde benzoïque, un ou plusieurs oses (surtout D-glucose) et l’acide cyanhydrique capable de provoquer de graves intoxications. À ce groupe appartient l’amygdaline , qui est formée de deux molécules de D-glucose combinées en un diholoside, le gentiobiose (tabl. 3, formule 4).
Hétérosides anthraquinoniques
De nombreuses teintures d’origine végétale sont des hétérosides, dont les plus importants sont ceux de la garance (Rubia tinctorum ). L’hydrolyse du principal hétéroside de la garance libère du D-glucose et du D-xylose (en combinaison disaccharidique) et la 1,2-dihydroxyanthraquinone ou alizarine. Celle-ci entre dans la composition de nombreuses peintures. C’est le premier colorant végétal dont la structure chimique fut déterminée, ce qui permit de le remplacer par un produit synthétique de meilleure qualité.
Une autre teinture (connue sous le nom de jaune indien) est préparée au Bengale en nourrissant le bétail de feuilles de mangues; le colorant, extrait des urines, est un hétéroside comprenant de l’acide D-glucuronique combiné à une molécule très voisine de l’alizarine, la 4,7-dihydroxyxanthone.
Hétérosides hétérocycliques oxygénés
La grande diversité des pigments des fleurs et des fruits dépend largement de l’existence de mélanges d’hétérosides et de caroténoïdes [cf. PIGMENTS]. Les couleurs bleues et certaines couleurs rouges sont dues à des hétérosides (anthocyanosides) dont les aglycones sont des anthocyanidines (cf. biogenèse des composés PHÉNOLIQUES).
Hétérosides sulfurés
La moutarde est préparée en broyant des graines de moutarde avec du sel, des épices et du vinaigre. De nombreuses variétés de graines de moutarde sont utilisées, mais toutes renferment un hétéroside où le D-glucose s’attache directement à un atome de soufre (tabl. 3, formule 5).
Autres hétérosides
Les tanins , dérivés de l’acide gallique et d’autres acides polyphénoliques, ne sont pas à proprement parler des osides, mais ils résultent de l’estérification, par ces acides, des fonctions alcooliques du glucose.
Les nucléosides , formés par l’union du ribose ou du désoxyribose avec des bases puriques et pyrimidiques, qui sont des produits de lyse partielle des acides nucléiques (cf. acides NUCLÉIQUES), se rattachent également aux hétérosides.
4. Métabolisme
Le L-glucose n’existant pas dans la nature, c’est au métabolisme du D-glucose que se ramène celui d’autres hexoses, tels que le D-mannose ou le D-galactose. On étudiera d’abord l’utilisation du glucose au niveau des tissus. Les transformations de cet ose empruntent, selon les circonstances, deux voies. La première ne fournit de l’anhydride carbonique qu’au-delà de son stade ultime: l’acide pyruvique, c’est la chaîne dite d’Embden-Meyerhof-Parnas (glycolyse ). La seconde produit de l’anhydride carbonique dès les premières étapes, c’est la chaîne des pentoses . On examinera ensuite le processus conduisant au glycogène, et d’une façon générale aux polysaccharides par l’intermédiaire des nucléotides-oses (UDP-glucose par exemple).
Glycolyse (voie des trioses)
Le glucose étant généralement stocké dans les cellules sous forme d’oligoholosides (saccharose) ou de polyholosides (amidon, glycogène), des réactions d’hydrolyse (catalysées par des enzymes) peuvent intervenir pour le libérer: par exemple, le saccharose est hydrolysé en glucose et fructose par action de l’invertase de la levure ou de l’ 見-glucosidase du suc intestinal; l’amidon en maltose par l’amylase, et le maltose en deux molécules de glucose par la maltase. Il faut remarquer que les ruminants ne synthétisant pas les cellulases, enzymes qui catalysent l’hydrolyse de la cellulose (principal constituant des tissus végétaux) en glucose, ne peuvent pas tirer l’énergie potentielle contenue dans le matériel végétal cellulosique (herbe, fourrage). Pour pallier ce désavantage d’un régime herbivore, ils vivent en symbiose avec certains micro-organismes, qui réalisent la dégradation du polysaccharide dans le rumen (ou panse).
Étapes
– Phosphorylation du glucose : L’utilisation du glucose fait intervenir, dès le premier stade, un processus de phosphorylation: le catabolisme du glucose se fait presque toujours sous forme de dérivés contenant au moins un groupement ester d’acide phosphorique. Le rôle fondamental joué par les dérivés phosphorylés a été établi par les travaux de O. Meyerhof, A. Harden, W. J. Young, entre 1900 et 1920.
La figure 2 montre que le glucose peut être directement phosphorylé par l’hexokinase, enzyme qui utilise une molécule d’ATP pour former du glucose-6-phosphate. D’autre part, le glycogène (ou l’amidon) peut être dégradé à partir des extrémités terminales (non-réductrices) des diverses chaînes par action des phosphorylases (la phosphorylase a étant la forme active de l’enzyme): une molécule de glucose est détachée de la chaîne du polysaccharide et transférée sur une molécule de phosphate pour former du glucose-1-phosphate. Ce processus très complexe (glycogénolyse) est contrôlé par des hormones: glucagon (produit par le pancréas), adrénaline (d’origine médullo-surrénale). Le glucose-1-phosphate est interconvertible en glucose-6-phosphate grâce à la phosphogluco-mutase.
– Sort du fructose-6-phosphate : En raison de l’équilibre qui existe entre les formes cyclique et linéaire, le glucose-6-phosphate est transformé en fructose-6-phosphate par la phosphofructo-isomérase, puis sous l’action de la phosphofructokinase, et d’une nouvelle molécule d’ATP, il est phosphorylé en fructose-1,6-diphosphate; cette réaction, énergétiquement irréversible, joue un rôle important dans la régulation du métabolisme cellulaire, l’enzyme étant inhibée par un excès d’ATP ou de citrate, et stimulé par l’AMP.
– Sort du fructose-1,6-diphosphate : Le fructose-1,6-diphosphate, sous sa forme linéaire, est scindé par l’aldolase (suivant une réaction inverse de l’aldolisation) en deux molécules de trioses-phosphates: la dihydroxy-acétone-phosphate et la D-glycéraldéhyde-3-phosphate. En raison de la formation de trioses, cette voie de dégradation, qui constitue la glycolyse, est aussi appelée «voie des trioses» (ou encore, voie de Embden – Meyerhof – Parnas).
– Oxydation du D-glycéraldéhyde-3-phosphate : Les deux trioses-phosphates sont en équilibre, grâce à la triosephosphate-isomérase. La dihydroxy-acétone-phosphate peut être transformée, soit en acide L- 見-glycérophosphorique (nécessaire à la synthèse des phospholipides), soit en une autre molécule de glycéraldéhyde-phosphate, déshydrogénée par un enzyme à groupe -SH, dont le coenzyme est le nicotinamide-adénine-dinucléotide (NAD+); cette déshydrogénation requiert du phosphate minéral, et il se forme un composé riche en énergie: l’acide glycérique-1,3-diphosphate (anhydride mixte de l’acide glycérique-3-phosphate et de l’acide phosphorique), ainsi qu’une molécule de NADH (coenzyme réduite).
– Formation de l’ acide pyruvique : L’hydrolyse enzymatique de cet anhydride mixte, en acide glycérique-3-phosphate, s’accompagne de la synthèse d’une mole d’ATP, au cours d’une réaction de «phosphorylation liée au substrat» (terme utilisé pour distinguer cette synthèse d’ATP de celle qui a lieu au niveau des mitochondries, au cours de l’«oxydation phosphorylante»).
L’acide glycérique-3-phosphate est isomérisé en acide glycérique-2-phosphate, puis déshydraté par l’énolase en phosphoénol-pyruvate (PEP), substance «riche en énergie» dont la déphosphorylation en acide pyruvique permet de synthétiser une nouvelle molécule d’ATP à partir d’une molécule d’ADP.
Bilan
Au cours de la glycolyse, une mole de glucose a été transformée en deux moles d’acide pyruvique. La formation de glucose-6-phosphate, puis de fructose-1,6-diphosphate, ont entraîné la consommation de deux moles d’ATP. En revanche, la formation d’acide glycérique-3-phosphate, puis l’utilisation du phosphoénol-pyruvate, ont permis de synthétiser deux moles d’ATP par mole de triose, c’est-à-dire quatre moles d’ATP par mole de glucose initial. Il existe donc un bénéfice de deux moles d’ATP par mole de glucose (trois moles d’ATP par mole du glycogène ou d’amidon) et, en outre, deux moles de NADH.
Sort de l’acide pyruvique
L’acide pyruvique (fig. 3) pourra:
1. Soit être réduit enzymatiquement en acide lactique, en utilisant l’hydrogène apporté sous forme de NADH au cours de la glycolyse. C’est ce qui se passe durant la contraction musculaire: une mole de glucose est transformée en deux moles d’acide L-lactique, avec apparition de deux moles d’ATP dont l’hydrolyse en ADP constitue la source énergétique de la contraction.
2. Soit être décarboxylé enzymatiquement en acétaldéhyde, dont l’hydrogénation (avec NADH) produit l’alcool éthylique. Ce phénomène est réalisé par la levure vivant en anaérobiose: c’est la fermentation alcoolique (cf. FERMENTATION, LEVURES).
Ces deux processus utilisent le glucose avec un rendement très faible, puisque 66 kilojoules sont récupérées, sur 7 065 kilojoules que peut théoriquement fournir la molécule de glucose.
3. Soit subir le phénomène de décarboxylation oxydative qui, par la mise en jeu d’un système enzymatique complexe, le transforme en acétyl-coenzyme A (CH3 漣CO 漣 SCoA) avec production d’une mole (par triose) de NADH qui vient s’ajouter à celle précédemment formée et non utilisée. À partir d’une mole de glucose, on obtient donc: deux moles d’ATP, quatre moles de NADH, et deux moles d’acétyl-coenzyme A. L’oxydation de l’hydrogène sous forme de NADH produit de l’énergie [cf. OXYDORÉDUCTIONS BIOLOGIQUES], ainsi que l’oxydation de l’acétyl-coenzyme A dans le cycle de Krebs [cf. MÉTABOLISME]. Cela augmente considérablement le rendement énergétique du catabolisme du glucose. Ce processus fonctionne dans la respiration : l’accepteur final de l’hydrogène est l’oxygène moléculaire.
Un organisme tel que la levure, capable de vivre en présence ou en absence d’oxygène, pourra donc, soit dégrader complètement le glucose en eau et gaz carbonique (respiration) soit le dégrader seulement au stade de produits du type alcool éthylique (fermentation). L’oxygène ralentit l’utilisation du glucose et assure sa dégradation poussée (effet Pasteur).
– L’acide pyruvique peut aussi servir à la production d’un acide aminé, l’alanine, et d’un 見-céto-acide plus complexe, l’acide oxaloacétique (indispensable au fonctionnement du cycle de Krebs, et précurseur de l’acide aspartique). Enfin, l’acétyl-coenzyme A peut servir de point de départ à la synthèse des stérols et à celle des acides gras, c’est-à-dire qu’en particulier, il permet la transformation réciproque des glucides en lipides.
Voie des pentoses
La glycolyse joue un rôle très important dans la production d’énergie, soit en aérobiose, soit en anaérobiose. Cependant, certains micro-organismes (notamment Acetobacter suboxydans ) ne possèdent pas tous les enzymes nécessaires pour que cette voie soit fonctionnelle, et d’autre part, certains inhibiteurs peuvent bloquer la voie des trioses sans interrompre la consommation du glucose. Ces observations ont conduit à mettre en évidence d’autres voies de catabolisme du glucose qui, pour la plupart, entraînent aussi sa phosphorylation immédiate.
– Oxydation du glucose-6-phosphate : Le glucose-6-phosphate peut être déshydrogéné en lactone de l’acide gluconique-6-phosphate par une déshydrogénase dont le coenzyme est le nicotinamide-adénine-dinucléotide-phosphate (NADP+); la lactone, par hydrolyse, fournit l’acide à chaîne ouverte (fig. 4).
– Oxydation décarboxylante de l’acide gluconique-6-phosphate : L’acide gluconique-6-phosphate est déshydrogéné par une seconde déshydrogénase à NADP+, au niveau de la fonction alcool secondaire en position 3. Il se forme ainsi un acide 廓-cétonique instable, qui perd spontanément une molécule de gaz carbonique en donnant un dérivé de cétose: le ribulose-5-phosphate.
– Les transformations du ribulose-5-phosphate : Le ribulose-5-phosphate peut être isomérisé en ribose-5-phosphate (utilisable pour la synthèse des acides nucléiques, constituants fondamentaux de la cellule), ou épimérisé en xylulose-5-phosphate. À partir de ces deux nouveaux pentoses, par intervention des enzymes transcétolase et transaldolase, des fragments ayant respectivement deux et trois atomes de carbone sont détachés des molécules de sucres cétoniques et fixés sur un sucre aldéhydique. Ces réactions conduisent à la formation de sédoheptulose-7-phosphate, d’érythrose-4-phosphate (nécessaire à la synthèse du noyau benzénique de certains acides aminés), de glycéraldéhyde-3-phosphate et de fructose-6-phosphate (pouvant être utilisés dans la voie des trioses). En dehors de la production de NADPH nécessaire à la synthèse des acides gras (à partir d’acétyl-coenzyme A), cette voie permet donc la formation d’érythrose et de ribose, métabolites indispensables au fonctionnement cellulaire.
Chez les êtres supérieurs, la voie des trioses et la voie des pentoses coexistent; leur importance relative est fonction de l’état physiologique et des conditions dans lesquelles se trouvent placées les cellules. Ainsi, dans le tissu lymphatique, la voie des pentoses semble très active. Dans les organismes chlorophylliens, le ribulose-5-phosphate est phosphorylé, par une kinase et l’ATP, en ribulose-1,5-diphosphate; ce dernier joue un rôle fondamental dans la fixation du gaz carbonique atmosphérique et sa transformation en glucose [cf. PHOTOSYNTHÈSE].
Voie du glucuronate-xylulose
Cette voie a été mise en évidence à la suite de la détection de L-xylulose dans le sang. En réalité, comme il ressort de l’examen de la figure 5, cette voie correspond davantage à un processus de métabolisation de l’acide glucuronique (susceptible d’être apporté par l’alimentation) qu’à une voie de métabolisation du glucose lui-même. Le L-xylulose formé est réduit en xylitol (pour lequel il n’existe plus de série stéréochimique: L-xylitol = D-xylitol). La réductase qui catalyse cette hydrogénation n’existe pas chez certains individus: le L-xylulose est alors éliminé dans l’urine, ce qui peut prêter à confusion avec l’élimination de glucose par les diabétiques lorsque la détection repose seulement sur la recherche d’un sucre réducteur dans l’urine.
Le xylitol est ensuite oxydé enzymatiquement en D-xylulose qui, après phosphorylation en D-xylulose-5-phosphate, peut être utilisé par la voie des pentoses.
Gluconéogenèse
La plupart des êtres vivants sont capables de synthétiser du D-glucose à partir de métabolites aussi simples que l’acétate. Ce processus constitue la gluconéogenèse, pouvant aller (chez l’homme en particulier) jusqu’au stade du glycogène. Ainsi l’acétate, sous la forme activée d’acétyl-coenzyme A, peut être incorporé dans l’acide oxaloacétique par le jeu du cycle de Krebs (fig. 3). En présence de GTP (guanosine-triphosphate) comme source d’énergie et d’une enzyme (phosphoénol-pyruvate-carboxykinase), l’oxaloacétate perd du gaz carbonique C2; il est transformé en phosphoénol-pyruvate (PEP).
Or, dans la chaîne qui va du glucose-6-phosphate au phosphoénol-pyruvate, toutes les réactions sont réversibles (et par suite susceptibles de fournir des produits de synthèse), sauf celle qui assure la transformation du fructose-6-phosphate en fructose-1,6-diphosphate (fig. 2). À ce stade intervient une enzyme spécifique de la synthèse, la fructose-diphosphatase, qui hydrolyse le groupe ester phosphorique en 1. Cette enzyme est stimulée par l’ATP et inhibée par l’AMP, ce qui assure son fonctionnement (et par suite la gluconéogenèse) lorsqu’il y a pléthore d’ATP (c’est-à-dire de réserves d’énergie immédiatement utilisables) dans la cellule. Le glucose-6-phosphate pourra être hydrolysé par la glucose-phosphatase en glucose libre.
Synthèse des polyholosides
Lorsque le glucose doit être transformé en glycogène (glycogénogenèse), le glucose-6-phosphate (provenant par exemple de la gluconéogenèse) est isomérisé par la phosphogluco-mutase en glucose-1-phosphate; ce dernier, en présence d’une enzyme du groupe des transférases, réagit avec l’UTP pour donner l’uridine-diphosphate de glucose (UDP-glucose: nucléotide constituant une forme générale de transport des sucres dans la synthèse d’oligo- ou de polyholosides). En particulier, une nouvelle transférase peut utiliser l’UDP-glucose pour fixer le résidu de glucose par une liaison glycosidique 見-1,4, à l’hydroxyle en 4 d’une unité glucose appartenant à une molécule de glycogène en voie de synthèse. Grâce à ce processus, une chaîne de glycogène s’allonge par additions successives d’unités glucose.
Lorsque la chaîne linéaire a atteint une certaine longueur, une enzyme « branchante» détache une partie de la molécule et la porte sur la fonction alcool primaire (en 6) d’une unité glucose, créant une liaison glycosidique 見-1,6, ce qui entraîne la ramification du polysaccharide. C’est de cette manière que le glycogène se reforme, en particulier dans le foie (fig. 6).
Il faut remarquer que le foie possède toutes les enzymes nécessaires à la synthèse du glycogène aussi bien à partir du glucose que de l’acide lactique, tandis que l’équipement enzymatique des cellules musculaires est limité aux enzymes qui assurent l’enchaînement de molécules de glucose (apportées par le sang) pour former le glycogène. Grâce à sa réserve de glycogène, le foie maintient la teneur du sang en glucose à une valeur à peu près constante (glycémie), de 70 à 100 mg pour 100 ml de sang chez l’homme.
La biosynthèse des polysaccharides chez les bactéries utilise une voie tout à fait semblable, mais le nucléotide diphosphate de transport est un ADP-ose (au lieu d’un UDP-ose). Chez les plantes, les deux types de nucléotides (avec leurs enzymes spécifiques) fonctionnent, mais la voie de synthèse prépondérante utilise les nucléotides à adénine.
La synthèse de polysaccharides extracellulaires fait intervenir une forme de transport (et d’activation) des oses plus lipophile que la forme nucléoside-diphosphate d’ose, étant donné la nécessité de traverser la membrane cytoplasmique. Les oses sont transformés en polyprényl-diphosphate d’ose, dans lesquels le sucre, lié par son hydroxyle glycosidique au pont diphosphate, se trouve fixé sur une longue molécule hydrophobe de nature terpénique. Un des transporteurs le plus fréquemment rencontrés est l’undécaprényl-phosphate, molécule en C55 avec onze ramifications méthyle.
Catabolisme avec interconversion
La figure 7 montre comment le catabolisme du D-mannose, du D-galactose et du D-fructose se ramène à celui du D-glucose. Pour être transformé en glucose, le galactose doit être incorporé dans un nucléotide dérivant de l’uridine. C’est sous la forme d’uridine-diphosphate de galactose (UDP-galactose) qu’une épimérase inverse la disposition des substituants sur le carbone 4 du sucre, conduisant à la formation d’UDP-glucose qui peut, ultérieurement, libérer du glucose-1-phosphate.
Encyclopédie Universelle. 2012.