ALCOOLS
Le mot alcool, de l’arabe al khoul ou al koh’l , désignait à l’origine une poudre très fine à base de stibine (sulfure d’antimoine). À ce mot s’attacha une idée de finesse et de subtilité, de sorte que les alchimistes l’appliquaient aussi bien à des poudres impalpables résultant d’une sublimation qu’aux principes volatils isolés par distillation. Paracelse nommait alcool vini celui qu’on recueillait dans la distillation du vin. À partir du début du XIXe siècle, ce dernier sens du mot alcool, employé sans qualificatif, devint à peu près exclusif. L’alcool de vin – dont le nom officiel est éthanol – avait donné lieu à un nombre important de transformations chimiques, et celles-ci présentaient de grandes analogies avec celles qui caractérisaient deux autres composés: l’alcool de bois, aujourd’hui appelé le méthanol, et l’alcool cétylique extrait du blanc de baleine. Progressivement, le mot alcool devint générique et désigna l’ensemble des composés dérivant d’un hydrocarbure par substitution, sur un carbone saturé, d’un hydrogène par un hydroxyle. Les alcools sont donc les composés portant le groupe fonctionnel hydroxyle 漣OH sur un carbone saturé (sp3) de la chaîne hydrocarbonée.
La nature nous fournit un grand nombre d’alcools, à fonction simple, soit sous la forme d’alcools proprement dits, soit sous forme de dérivés, notamment d’esters: les alcools de fermentation, les huiles essentielles, le glycérol des lipides, le cholestérol des tissus animaux, les alcools laurique en C12 de la noix de coco, cétylique en C16 du blanc de baleine et cérylique en C26 de la cire de Chine. Les sucres sont des polyalcools aldéhydiques ou cétoniques.
1. Divers types d’alcools et leur nomenclature
On appelle classe d’un alcool le degré de substitution du carbone saturé qui porte la fonction hydroxyle: au carbone primaire, monosubstitué, correspond l’alcool primaire R 漣CH2OH, au carbone secondaire, bisubstitué, l’alcool secondaire RR CHOH et au carbone tertiaire, trisubstitué, l’alcool tertiaire RR R COH. Le méthanol, CH3OH, est seul de son espèce. On distingue également les alcools dont la fonction hydroxyle est portée par un carbone sp3 lié à une chaîne insaturée: alcools allyliques

qui présentent des propriétés spécifiques.
La fonction alcool peut aussi se trouver fixée sur un squelette porteur d’une ou de plusieurs autres fonctions: diols ou glycols, triols ou glycérols, etc.; acides-alcools, aldéhydes-alcools ou aldols, cétones-alcools ou cétols, amines-alcools ou aminoalcools, etc.
La nomenclature officielle de l’I.U.P.A.C. (International Union of Pure and Applied Chemistry) forme le nom d’un alcool en remplaçant l’e final du nom de l’hydrocarbure dont il dérive par le suffixe -ol . Lorsque la chaîne est ramifiée, le nom de l’alcool dérive de celui de la chaîne la plus longue qui porte la fonction, et le numérotage donne la priorité du plus petit nombre au carbone hydroxylé. Une nomenclature courante, due à Kolbe, fait dériver le nom des alcools de celui du plus simple, le méthanol appelé carbinol, en dénommant le ou les groupes qui remplacent l’hydrogène de ce carbinol. Enfin, une nomenclature usuelle les désigne comme alcools alkyliques.
2. Préparations et modes de formation
Fermentation
La fermentation alcoolique des jus sucrés sous l’action de micro-organismes tels que les levures et les bactéries est une source d’alcools. L’éthanol est fabriqué industriellement par fermentation, sous l’action du complexe enzymatique zymase, présent dans la levure de bière, de mono- et disaccharides comme le glucose, le fructose, le saccharose, etc. provenant de l’hydrolyse de polysaccharides tels que l’amidon, la fécule et la cellulose ou de jus sucrés naturels comme le sucre de canne et les mélasses de sucrerie. Le butanol-1 est obtenu en mélange avec l’acétone par fermentation de sucres en C6 tels que le glucose et le fructose, en C5 (xylose), de disaccharides comme le saccharose et le lactose ou de polysaccharides (amidon), en présence d’une bactérie, le Clostridium acetobutylicum .
Synthèses industrielles
Le méthanol, qui est l’une des plus importantes matières premières industrielles de synthèse, est fabriqué par hydrogénation catalytique du monoxyde de carbone: le gaz de synthèse (CO + 2H2) obtenu soit par oxydation du méthane, soit par gazéification du charbon est traité à 250 0C et sous 50 bars par un catalyseur à base de CuO, ZnO, Al23:

L’éthanol est fabriqué industriellement par hydratation de l’éthylène, soit indirectement par absorption par l’acide sulfurique à 95 p. 100 puis hydrolyse des esters sulfuriques d’éthyle, soit directement par passage du mélange gazeux d’éthylène et d’eau sur un catalyseur d’acide phosphorique sur silice (réactions 1).
L’hydratation indirecte du propylène par l’acide sulfurique à 75 p. 100, puis hydrolyse, ou directe sur acide phosphorique, conduit à l’isopropanol (réactions 2).
Le butane-1-ol est fabriqué par deux voies: soit l’hydroformylation du propène ou synthèse OXO sur catalyseur au cobalt ou au rhodium, suivie de l’hydrogénation du butanal, avec coproduction d’isobutanol (réaction 3); soit l’aldolisation de l’éthanal, la crotonisation de l’aldol puis l’hydrogénation du crotonaldéhyde:

Le butane-2-ol, résulte de l’hydratation indirecte de la coupe C4 de vapocraquage, débarrassée du butadiène et de l’isobutène: le butène-1 et les butènes-2 sont hydratés sélectivement en alcool secondaire (réactions 4). Le 2-méthylpropanol ou tertiobutanol est fabriqué par hydratation indirecte de l’isobutène contenu dans la coupe C4 débarrassée du butadiène: des n -butènes, c’est le plus réactif, et l’emploi d’un acide sulfurique dilué à 55 p. 100 permet de l’absorber sélectivement (réaction 5). L’isobutène peut aussi être extrait d’une manière sélective en lui additionnant du méthanol en catalyse acide, pour former l’éther de méthyle et de tertiobutyle (M.T.B.E.). L’époxydation du propène par l’hydroperoxyde de tertiobutyle donne le tertiobutanol, c’est le procédé Oxirane (réaction 6).
Deux séries d’alcools supérieurs sont fabriquées: un premier groupe d’alcools primaires en C6-C9 utilisé pour la synthèse de plastifiants (esters) et un second en C12 et plus pour la fabrication de détergents. Un procédé général appelé Aldox est utilisé pour la synthèse des alcools du premier groupe: il fait intervenir l’hydroformylation d’une 見-oléfine, l’aldolisation de l’aldéhyde obtenu (doublement de la chaîne carbonée), sa crotonisation et une hydrogénation:

Un second procédé permet la synthèse d’alcools primaires à chaîne linéaire à partir d’éthylène: il réalise l’oligomérisation de l’éthylène sur catalyseur de triéthylaluminium, l’oxydation du trialkylaluminium puis l’hydrolyse de l’alcoolate. Le mélange d’alcools obtenus peut contenir des chaînes de C4 à C16 avec un maximum de population en C12:

Des alcools à longue chaîne ou alcools gras sont obtenus par méthanolyse de corps gras naturels tels que les graisses animales ou végétales puis hydrogénolyse des esters méthyliques des acides gras (réaction 7).
Synthèses de laboratoire
Hydrolyse de dérivés fonctionnels monovalents
L’eau, réactif nucléophile, ou sa base conjuguée H- qui est présente dans des bases alcalines NaOH et KOH, réagit sur les substrats porteurs de groupements fonctionnels comme les halogénures et les esters. La réaction est une substitution nucléophile, généralement bimoléculaire (SN2), sauf dans le cas des halogénures tertiaires et des esters d’alcools tertiaires où le mécanisme peut être unimoléculaire (SN1). Dans le cas où le réactif est une base forte, la substitution peut être accompagnée d’une élimination et conduire à un éthylénique. Ce mode de préparation ne présente d’intérêt que si le dérivé fonctionnel, soumis à l’hydrolyse, n’est pas lui-même issu de l’alcool. C’est ainsi que l’on prépare industriellement l’alcool allylique par hydrolyse à la soude du chlorure d’allyle, obtenu par chloration radicalaire du propène:
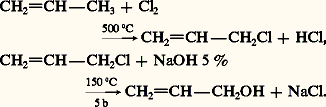
La saponification d’esters d’alcools gras naturels, présents dans les cires, libère l’alcool en formant un sel alcalin de l’acide gras (savon). Par exemple, le traitement par la soude du palmitate de cétyle, principal constituant du blanc de baleine, soustrait l’alcool cétylique et forme le palmitate de sodium:

L’alcoolyse des esters, en catalyse acide, permet de libérer l’alcool de l’ester:

L’hydrolyse des esters sulfuriques d’alkyle, obtenus par addition de l’acide sur un alcène, est une réaction de ce type.
Hydratation des alcènes
Cette réaction, pratiquée industriellement pour la fabrication des alcools en C2, C3 et C4, conduit essentiellement aux alcools secondaires et tertiaires (sauf pour l’éthanol) du fait de l’orientation de l’addition [cf. ALCÈNES]. L’hydratation indirecte d’un alcène terminal par la suite de réactions d’hydroboration, d’oxydation et d’hydrolyse permet d’obtenir l’alcool primaire (réaction 8).
Hydrogénation de dérivés carbonylés
La réduction des aldéhydes et des cétones en alcools primaires et secondaires peut être réalisée par l’action de l’hydrogène en présence de catalyseur métallique comme le platine Pt et le nickel Ni:

Lorsque la chaîne du dérivé carbonylé comporte un groupe réductible par l’hydrogène moléculaire, une liaison éthylénique par exemple, la réduction de la seule fonction carbonyle peut être réalisée par l’action d’un hydrure métallique complexe comme LiAlH4, qui est soluble dans l’éther et décomposé par l’eau, NaBH4 soluble dans l’eau sans décomposition. La réaction suivante montre, sur l’exemple du crotonaldéhyde, l’hydrogénation sélective de la fonction carbonyle, avec conservation de la fonction éthylénique, pour former l’alcool crotylique alors que l’hydrogénation catalytique sur nickel du même aldéhyde insaturé conduit au butanol-1:
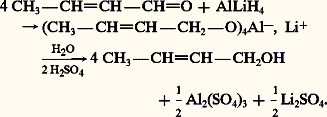
L’hydrure complexe de lithium et d’aluminium est susceptible de réduire en alcools primaires les acides carboxyliques qui résistent à la plupart des agents d’hydrogénation: l’acide pivalique est ainsi transformé en alcool néopentylique (réaction 9). Les esters sont également réduits en alcools par AlLiH4 ou par action d’un excès de sodium en présence d’éthanol absolu (Bouveault et Blanc): ce dernier cas est appliqué aux cétones, les aldéhydes étant sensibles aux milieux basiques:

Les esters d’acides gras sont réduits industriellement en alcools gras par l’hydrogène sur catalyseur de chromite de cuivre Cr23, CuO (Adkins): la réaction suivante est l’exemple du laurate de méthyle (obtenu par méthanolyse de la laurine, ou trilaurate de glycérol, extraite de la noix de coco) qui est transformé en alcool laurique:

Une autre réaction spécifique de l’hydrogénation des fonctions carbonyle est l’échange fonctionnel carbonyle-alcool, de Meerwein, Ponndorf, Verley: le dérivé carbonylé à réduire est traité par un alcool secondaire, l’isopropanol, en présence d’une quantité catalytique d’isopropylate d’aluminium. L’échange fonctionnel est réversible et l’équilibre est déplacé vers la formation de l’alcool recherché par élimination de l’acétone formée, qui est le constituant le plus volatil. Cette méthode respecte les autres fonctions réductibles (halogène, nitro, double liaison) [réaction 10].
Addition d’organométalliques sur les fonctions carbonyle
Les organomagnésiens s’additionnent sur le méthanal ou sur un aldéhyde ou encore sur une cétone pour donner respectivement des alcools primaire, secondaire et tertiaire. Cette méthode de préparation est très générale.
Les chlorures d’acides et les esters réagissent sur les organomagnésiens en donnant des cétones, non isolées, qui sont transformées en alcools tertiaires. L’oxyde d’éthylène conduit aux alcools primaires.

L’acétylène s’additionne sur les aldéhydes et les cétones par catalyse basique ou en présence d’acétylure de cuivre et d’argent. Cette réaction est appliquée industriellement à la préparation du butyne-1,4-diol qui, par hydrogénation, conduit au butane-1,4-diol:

3. Propriétés physiques
La présence de la fonction hydroxyle entraîne, pour les alcools, un ensemble de propriétés physiques particulières: les molécules sont polaires (tabl. 1) et associées par des liaisons hydrogène (fig. 1). Surtout sensible pour les premiers termes, cette association intermoléculaire se traduit par un ensemble de propriétés caractéristiques. Moins spectaculaire que pour l’eau, bien que de même nature, elle est importante pour les alcools primaires, notable pour les secondaires et faible pour les tertiaires. Elle diminue lorsque la température s’élève.
Une première conséquence de cette association est une température d’ébullition des alcools notablement supérieure à celle des alcanes de même masse moléculaire (fig. 2), avec une anomalie dans l’évolution de la volatilité des alcools primaires linéaires: alors que dans la série des n -alcanes (liquides idéaux) l’écart entre les températures d’ébullition de deux homologues consécutifs diminue lorsque la chaîne s’allonge, dans le cas des alcools cet écart, du moins jusqu’au terme en C10, est sensiblement constant et voisin de 19 0C. Cette anomalie résulte d’une compensation, presque rigoureuse, entre l’effet de l’allongement de la chaîne et la diminution de l’association avec l’élévation de la température. Cette association explique l’écart notable des températures d’ébullition d’alcools primaires, secondaires et tertiaires isomères ainsi que la proximité de celles des alcools C2 primaire, C3 secondaire et C4 tertiaire.
L’affinité pour l’eau des alcools, notamment des premiers termes, entraîne l’existence d’une azéotropie homogène ou hétérogène, mais toujours positive. L’azéotrope eau-éthanol bout seulement à 0,25 0C plus bas que l’éthanol pur (respectivement 78,15 0C et 78,40 0C); il renferme environ 5 p. 100 d’eau en poids et est désigné sous le nom d’alcool à 95o. Le pourcentage d’eau dans les azéotropes augmente avec la masse moléculaire de l’alcool.
Les températures de fusion des alcools ne présentent pas de loi simple d’évolution en fonction de la masse moléculaire, du moins pour les premiers termes. Pour les alcools primaires linéaires plus longs, on observe une progression voisine de celle observée pour les alcanes de même chaîne. Les alcools tertiaires symétriques cristallisent plus facilement que leurs isomères primaires et secondaires. Par exemple, le butanol tertiaire fond à 25,5 0C, alors que les isomères n -primaire, iso-primaire, secondaire fondent respectivement à 漣 79,9 0C, 漣 108 0C et 漣 114,7 0C. De nombreux alcools naturels, appartenant à la série terpénique, sont solides à la température ordinaire (l (face=F0019 漣) menthol F 43 0C; d ,l bornéol F 208 0C).
Du fait de leur constante diélectrique relativement élevée (tabl. 1), les alcools en C1, C2 et C3 sont des solvants protoniques polaires pour les espèces ioniques. Leur capacité de former des associations par liaison hydrogène leur permet aussi de dissoudre des molécules à caractère acide et basique; c’est pourquoi ils sont couramment employés en synthèse organique. Ils sont, ainsi que le tertiobutanol, miscibles à l’eau en toute proportion. Les autres butanols restent solubles mais non miscibles, et la solubilité dans l’eau diminue lorsque la masse moléculaire augmente, puis elle devient négligeable à partir de C10; en revanche, la liposolubilité augmente.
Les termes simples tels que le méthanol et l’éthanol dissolvent facilement certains sels de métaux à caractère faiblement ionique: halogénures de lithium, chlorure mercurique, chlorure ferrique; ces solutions ne sont pas ionisées. Les sels à caractère ionique ont une solubilité variable en fonction de leur nature: les chlorures de sodium et de potassium sont très peu solubles, les iodures et les nitrites le sont; ces solutions restent peu ionisées et renferment vraisemblablement des paires d’ions solvatées. La soude et la potasse se dissolvent aisément alors que la baryte Ba(OH)2, les sulfates et carbonates sont totalement insolubles. L’éthanol, en particulier, est souvent utilisé pour éliminer l’un de ces sels d’une préparation organique. Les sucres sont bien moins solubles dans l’éthanol que dans l’eau.
Transparents dans l’ultraviolet et le visible, les alcools présentent, en absorption infrarouge, des bandes caractéristiques de la vibration d’élongation de la liaison O 漣H. Lorsque l’éthanol est en phase vapeur ou en solution très diluée dans un solvant apolaire comme CCl4, la bande 益O-H est fine et située à 3 650 cm-1: la molécule n’est pas associée. Si la concentration augmente, et a fortiori pour l’alcool pur, une seconde bande large apparaît autour de 3 350 cm-1 qui correspond à la vibration d’élongation de la liaison O 漣H de molécules associées par liaison hydrogène. L’apparition de cette bande à environ 300 cm-1 vers les basses fréquences correspond à un «alourdissement» de l’atome d’hydrogène, et son élargissement au fait que les molécules d’alcool sont associées en agrégats de taille et de forme variables, correspondant à des liaisons hydrogène de nature variée, dont les fréquences de vibration se situent dans un intervalle de plusieurs centaines de cm-1 autour d’une valeur centrale de 3 350 cm-1. De l’intensité relative des bandes 益O-H libre et 益O-H associé, on peut estimer le degré d’association de l’alcool.
Les propriétés organoleptiques et physiologiques des alcools varient avec leur nature. Le méthanol, l’éthanol et l’isopropanol, bien purs, sont à peu près inodores; les deux derniers, l’éthanol surtout, sont les excipients les plus courants des parfums naturels ou synthétiques. Les alcools tertiaires simples possèdent une odeur camphrée. Les premiers termes des alcools éthyléniques sont agressifs. Par contre, parmi les termes moyens se rencontrent de nombreux parfums naturels ou artificiels: géraniol, linalol, citronellol, bornéol, etc. (tabl. 2). Ils sont utilisés en parfumerie ainsi que leurs esters.
À des doses raisonnables, l’éthanol, présent dans les vins, cidres, poirés, bières, etc., et dans les spiritueux, n’est pas toxique; sa valeur stimulative est très contestée. À haute dose, il provoque l’ivresse; à dose exagérée et répétée, l’alcoolisme et la cirrhose. Toutefois, il est bien établi que les effets durables qu’on lui attribue sont dus en partie aux impuretés qui l’accompagnent dans les spiritueux: alcools supérieurs, aldéhydes ou cétones auxquels ils doivent leur arôme.
Le méthanol, parfois employé dans la confection frauduleuse des spiritueux, est beaucoup plus toxique. On lui doit de nombreux accidents mortels et des cas de cécité. Les alcools dit de «fusel», alcools supérieurs de fermentation, présents dans les eaux-de-vie mal rectifiées, sont également dangereux.
4. Propriétés chimiques
Structure électronique
Compte tenu de la grande électronégativité de l’oxygène, la molécule d’alcool connaît, au niveau du groupe fonctionnel, une importante polarisation des liaisons C 漣礪漣 O et O 漣麗漣 H. Celle-ci induit, dans le squelette hydrocarboné, une polarisation monotone des liaisons adjacentes, qui s’amortit dès la troisième liaison (effet induit ou effet de champ). Les réponses, à cette induction, des liaisons issues du carbone sp3 porteur du groupe OH, sont différentes en fonction du degré de substitution de cet atome: en effet, le carbone sp3 des alcanes est moins électronégatif que l’hydrogène, de sorte que la liaison C(sp3) 漣H est naturellement polarisée du carbone ( 嗀+) vers l’hydrogène ( 嗀-). La situation électronique des alcools dépend ainsi de leur classe, l’effet de champ de l’oxygène fonctionnel étant mieux relayé par la chaîne carbonée d’un alcool tertiaire que par celle d’un primaire.
Réactions mettant en jeu la liaison OH
Acidité, basicité
La fonction alcool présente à la fois un caractère acide et un caractère basique. Son acidité se manifeste lorsqu’il est mis au contact d’une base: le proton est cédé à cette dernière et l’anion alcoolate formé ainsi que l’acide conjugué de la base se trouvent en équilibre avec l’alcool non dissocié:

La basicité de l’alcool se manifeste lorsqu’il est mis en présence d’un acide protonique: il fixe le proton de ce dernier, et le cation alkyloxonium formé, ainsi que la base conjuguée de l’acide se trouvent en équilibre avec l’alcool non protoné:
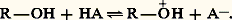
Étant à la fois acide et base, l’alcool pur se trouve en équilibre avec son acide et sa base conjugués:

Cet équilibre d’autodissociation de l’alcool est très déplacé vers la gauche, plus encore que celui de l’eau: le produit ionique du méthanol est de 10-17, alors que celui de l’eau est de 10-14.
En solution dans l’eau, les alcools sont donc faiblement dissociés selon l’équilibre:

dont la constante varie de 10-16 pour les alcools primaires à 10-18 pour les tertiaires.
Cette plus forte acidité des alcools primaires résulte de la meilleure dispersion de la charge négative de l’anion alcoolate sur le squelette hydrocarboné, qui entraîne une meilleure solvatation de cet anion.
La basicité des alcools est légèrement inférieure à celle de l’eau, et l’acide conjugué de l’alcool est d’autant mieux stabilisé par solvatation que sa charge positive est mieux dispersée sur le squelette hydrocarboné, ce qui entraîne une basicité décroissante dans l’ordre: alcool tertiaire 礪 secondaire 礪 primaire 礪 méthanol.
Formation et réactions d’alcoolates
Les dérivés métalliques d’acides plus faibles que les alcools réagissent avec ces derniers en formant des alcoolates: l’amidure de sodium, l’hydrure de sodium, le butyllithium et le bromure de méthylmagnésium réagissent avec l’éthanol en formant, d’une manière pratiquement totale, l’éthylate métallique correspondant (réactions 11).
Les métaux alcalins, l’aluminium amalgamé réagissent sur les alcools en formant les alcoolates métalliques correspondants, avec dégagement d’hydrogène:

Les alcoolates d’aluminium et de magnésium ne sont pas ionisés en solution alcoolique, les alcoolates alcalins le sont partiellement (paires d’ions solvatées). Ce sont des bases relativement fortes qui sont utilisées comme catalyseurs dans de nombreuses réactions (cf. ester et synthèses ACÉTYLACÉTIQUES). Les alcoolates sont complètement hydrolysés si l’eau est en grand excès; en proportions stœchiométriques, l’hydrolyse de l’éthylate de sodium est limitée à 70 p. 100, ce qui permet la fabrication de l’éthylate de sodium à partir de l’alcool absolu en grand excès et de la soude:

on déplace l’équilibre en éliminant l’eau sous forme d’azéotrope à 95 p. 100 d’alcool.
Les alcoolates sont des réactifs meilleurs nucléophiles que les alcools, ils interviennent dans des réactions d’éthérification. Agissant sur un halogénure, un sulfonate ou un sulfate d’alkyle, ils conduisent aux éthers (Williamson, 1850). La réaction de substitution S2 est beaucoup plus rapide que celle des alcools correspondants; elle peut être accompagnée d’une élimination E2 conduisant aux alcènes. Cette réaction secondaire est d’autant plus probable que l’alcool est tertiaire et que la température de réaction est élevée:

Les alcoolates primaires et secondaires sont également des réducteurs, comme dans l’échange fonctionnel alcool/aldéhyde-cétone catalysé par l’isopropylate d’aluminium: c’est la réduction de Meerwein-Ponndorf-Verley (réaction 10). Ce mécanisme fait intervenir un transfert d’hydrure dont le moteur est le passage du doublet libre de la fonction alcoolate en doublet 神 carbonyle (réaction 12).
Les alcoolates métalliques qui dérivent d’alcools porteurs en 廓 d’un groupe susceptible de stabiliser un carbanion (> C 略O, 漣C2R, 漣2, 漣S2) subissent plus ou moins facilement la fragmentation conduisant à ce dernier sous l’action d’une élévation de température (réaction 13).
Estérification
Bien que plusieurs mécanismes entrent en action dans l’estérification des acides carboxyliques par les alcools, le plus important d’entre eux fait intervenir la rupture de la liaison O 漣H. Cette réaction, très lente en l’absence de catalyseur, est fortement accélérée par les ions H+; sa limite à l’équilibre dépend de la classe de l’alcool: à partir d’un mélange stœchiométrique d’acide carboxylique et d’alcool, elle atteint 73 p. 100 pour le méthanol, 66 p. 100 pour les alcools primaires, 63 p. 100 pour les secondaires et moins de 10 p. 100 pour les tertiaires.
Le mécanisme le plus important correspond à l’activation nucléophile de l’acide par protonation, attaque de l’acide activé par l’alcool dans une étape d’addition nucléophile, trans-protonation suivie de l’élimination intramoléculaire du bon groupe partant H2O (réaction 14). Cette réaction d’estérification, réalisée avec un diacide et un glycol primaire, conduit aux polyesters utilisés pour la fabrication de fibres, de films, de résines:

Dans le cas d’un alcool tertiaire, un mécanisme différent fait intervenir la rupture de la liaison C 漣O. Étant plus basique, il se protone et subit une hétérolyse unimoléculaire qui conduit à un cation carbénium tertiaire. Ce dernier, fortement électrophile, attaque l’acide carboxylique pour former l’ester:

Les chlorures d’acides et les anhydrides d’acides réagissent plus rapidement avec les alcools primaires et secondaires en formant des esters:

Dans le cas des alcools tertiaires, il peut se produire une réaction parallèle de chloruration. Les acides minéraux faibles comme B(OH)3, H2 réagissent d’une manière analogue avec les alcools primaires; la réaction est également réversible.
Réactions d’addition
Les alcools s’additionnent aux composés insaturés polaires, l’oxygène jouant le rôle d’un nucléophile et l’hydrogène celui d’un électrophile.
Les dérivés carbonylés, aldéhydes et cétones additionnent les alcools primaires, en catalyse acide, pour donner des acétals [cf. ACÉTALS]:

Les époxydes réagissent, en catalyse acide, avec les alcools primaires en formant des éthers de glycols:

Les isocyanates forment, avec les alcools primaires, des carbamates appelés uréthannes: cette réaction est utilisée pour l’identification des alcools par la détermination de la température de fusion du phényluréthanne. Ce dernier, qui est un solide bien cristallisé, résulte de l’addition de l’alcool à l’isocyanate de phényle (réaction 15). La polyaddition d’un di-isocyanate à un glycol primaire ou secondaire conduit à un polyuréthanne qui est un polymère aux multiples usages: mousses souples, objets moulés, vernis polymérisables.
Réactions mettant en jeu la liaison C size=5漣O
Les acides halogénés attaquent les alcools en formant réversiblement des halogénures d’alkyle. Les réarrangements de Wagner-Meerwein du squelette carboné peuvent se produire chez les alcools tertiaires encombrés en 見 de la fonction. Dans le cas des alcools tertiaires, secondaires ou 見廓 insaturés, le mécanisme est normalement unimoléculaire (SN1) et fait intervenir un cation carbénium; ce dernier peut se réarranger en formant un cation plus stable ou, s’il est allylique, il offre au nucléophile halogénure plusieurs sites réactionnels. La bromuration du méthyltertiobutylcarbinol donne exclusivement le bromure de diméthylisopropylcarbinyle (réaction 16). De même, la chloruration du méthylvinylcarbinol conduit à un mélange des trois chlorures allyliques isomères (réaction 17). L’acide chlorhydrique, moins réactif que les acides bromhydrique et iodhydrique, peut être activé par un acide de Lewis comme ZnCl2. La réaction de ce superacide, avec les alcools, permet de déterminer leur classe: à température ordinaire, l’alcool tertiaire réagit très rapidement, le secondaire demande plusieurs minutes et le primaire nécessite un chauffage (test de Lucas).
Les oxacides forts comme H2S4, NH3 forment réversiblement à froid des esters avec les alcools primaires à température modérée (face=F0019 麗 130 0C). En présence d’un excès d’alcool, l’ester sulfurique formé est alcoolysé en éther; à température plus élevée (150 0C), il subit une élimination qui conduit à l’alcène. Les alcools tertiaires subissent essentiellement une déshydratation:

Les chlorures d’acides carboxyliques réagissent avec les alcools tertiaires en donnant simultanément un ester et un chlorure. L’attaque nucléophile de la fonction carbonyle, par l’alcool, aboutit à un intermédiaire qui peut soit éliminer un proton et conduire à l’ester, soit subir une hétérolyse formant un cation carbénium tertiaire et aboutir au chlorure d’alkyle (réaction 18).
Les chlorures d’acides minéraux, comme l’oxychlorure de phosphore OPCl3 et le chlorure de thionyle OSCl2, transforment les alcools en chlorure d’alkyle. Dans le cas du chlorure de thionyle, la réaction est stéréospécifique et se produit avec rétention de configuration; le chlorosulfite d’alkyle formé initialement se fragmente selon un mécanisme S1 de substitution nucléophile interne (réaction 19).
L’élimination d’une molécule d’eau (déshydratation) se produit très rapidement, en catalyse acide et à température modérée, pour des alcools tertiaires; elle est plus lente pour les secondaires et nécessite, pour les primaires, une température plus élevée (150 0C). La plupart des alcools se déshydratent à une température supérieure à 300 0C, en présence de catalyseurs solides comme la silice ou l’alumine, en donnant des alcènes, accompagnés parfois d’éthers si la température est plus modérée.
Réactions de déshydrogénation et d’oxydation
Le méthanol, les alcools primaires et secondaires subissent une déshydrogénation sous l’action de catalyseurs comme le nickel, le platine, le cuivre réduits. Cette réaction, réalisée en phase gazeuse, est réversible. On peut la rendre totale en la combinant à une oxydation ménagée par l’air. Il se forme un dérivé carbonylé: le méthanal dans le cas du méthanol, un aldéhyde dans le cas d’un alcool primaire, une cétone dans celui d’un alcool secondaire (réactions 20). Les alcools tertiaires ne possédant pas d’hydrogène sur le carbone porteur de la fonction ne subissent pas de déshydrogénation, tout au plus sont-ils déshydratés en alcènes dans les mêmes conditions. Le méthanal est fabriqué industriellement par oxydéshydrogénation du méthanol par un défaut d’air sur catalyseur d’argent à 680 0C. Un procédé de fabrication de l’éthanal réalise la déshydrogénation de l’éthanol sur catalyseur au cuivre, activé par le zinc, à 280 0C.
Le passage de l’alcool au dérivé carbonylé peut être réalisé à froid par oxydation en phase liquide. L’oxydant usuel est l’acide chromique H2Cr4 ou le bichromate de sodium Na2Cr27 et l’acide sulfurique. Le mécanisme de cette réaction fait intervenir la formation d’un chromate d’alkyle suivie d’une élimination inter ou intramoléculaire au cours de laquelle le chrome subit une réduction du degré d’oxydation 6(H2Cr4) au degré d’oxydation 4(H2Cr3) qui se dismute en donnant du chrome III et du chrome VI (réactions 21). Dans ces conditions, l’aldéhyde résultant de l’oxydation d’un alcool primaire est distillé au fur et à mesure de sa formation pour éviter une oxydation ultérieure en acide.
Vis-à-vis d’oxydants plus puissants comme le permanganate de potassium ou l’acide nitrique à 80 0C, les trois classes d’alcool réagissent spécifiquement. Les alcools primaires sont transformés en acide à même nombre d’atomes de carbone; en effet, la plupart des acides sont inoxydables dans ces conditions. Les alcools secondaires sont oxydés en cétones qui, si on insiste, subissent une rupture oxydante en un mélange d’acides et éventuellement de cétones peu oxydables; l’acétone résiste à cette oxydation ultérieure.
Les résultats sont plus complexes si les radicaux qui flanquent le groupe 漣CHOH 漣 sont ramifiés; seul l’isopropanol n’est pas dégradé. Les alcools tertiaires sont peu oxydables en milieu neutre; en milieu acide, seuls les alcools non déshydratables, comme (C6H5)3COH, résistent aux oxydants. Les autres sont déshydratés sous l’action catalytique de l’acide, et l’alcène résultant est très oxydable. Il se forme un glycol qui subit un clivage oxydant (réaction 22).
Une oxydation sélective des alcools primaires et secondaires peut être réalisée par un échange fonctionnel catalysé par le tertiobutylate d’aluminium. C’est la réaction d’Oppenauer, inverse de la réaction de Meerwein-Ponndorf-Verley (réaction 10); l’alcool à oxyder est mis en présence d’un large excès d’acétone (rapport molaire 1/50) et du catalyseur. L’équilibre ainsi déplacé par action de masse permet la transformation, pratiquement totale, de l’alcool en aldéhyde ou cétone.
Encyclopédie Universelle. 2012.