AMMONIAC
L’ammoniac est surtout connu par sa synthèse qui, en apportant une solution au problème de la fixation de l’azote de l’air, a révolutionné l’industrie chimique, ouvrant la voie à une technique nouvelle: celle des opérations sous pression. Il constitue maintenant l’un des produits chimiques dont le tonnage de production est très important, car, avec l’ammoniac, la chimie offre à l’homme l’une de ces possibilités étonnantes dont il peut se servir pour le bien ou le mal: constituant et matière première de la plupart des engrais azotés, il sert également à fabriquer l’acide nitrique, lui-même utilisé dans la fabrication de la plupart des explosifs.
Historique
On connaît depuis l’Antiquité le «sel ammoniac», c’est-à-dire le chlorure d’ammonium NH4Cl, dont le nom viendrait de celui du dieu égyptien Amon et qui, initialement obtenu par sublimation, en décomposant par la chaleur la fiente de chameau, était importé d’Orient.
En chauffant ce «sel» avec de la chaux, plusieurs alchimistes constatèrent la formation d’un gaz suffocant, mais ce fut J. Priestley qui, en 1774, l’isola en le recueillant sur la cuve à mercure et lui donna le nom «d’air alcalin». Le nom d’ammoniaque par lequel on désigne aujourd’hui la solution aqueuse du gaz ammoniac , fut proposé par T. Bergmann. Priestley avait été amené à considérer ce gaz comme un composé d’azote et d’hydrogène; cela fut confirmé par C. L. Berthollet, qui, en 1785, en fit l’analyse et indiqua sa composition; celle-ci correspond à la formule NH3, qui fait de ce produit le plus simple des hydrures d’azote.
Jusqu’au milieu du XIXe siècle on a extrait l’ammoniac des eaux-vannes, dans lesquelles il se forme par décomposition de l’urée: après 1850, on l’a obtenu comme sous-produit de l’industrie du gaz. La carbonisation de la houille donne lieu en effet à la formation d’ammoniac, qui se condense sous forme d’eaux ammoniacales avec les goudrons.
Les gaz de fours à coke contiennent aussi de l’ammoniac que l’on fixa généralement, lorsqu’on entreprit leur traitement, à l’état de sulfate d’ammonium, utilisé comme engrais ; on s’était en effet rendu compte, entre-temps, de l’importance de l’ammoniac comme constituant des engrais azotés, et c’est cette question des engrais qui devait révolutionner la préparation de l’ammoniac et aussi l’industrie chimique.
C’est dans la seconde moitié du XIXe siècle que s’est posé le problème de la fixation de l’azote de l’air. Devant la crainte de la famine mondiale prédite par l’économiste anglais R. Malthus, on a entrepris l’exploitation agricole intensive grâce à l’utilisation d’engrais chimiques. On eut recours initialement, comme engrais azotés, aux nitrates du Chili; mais craignant que leurs gisements ne soient trop rapidement épuisés, on a estimé nécessaire de faire appel à cette source intarissable constituée par l’azote atmosphérique, qu’on a cherché à fixer à l’état de composés susceptibles d’être transformés en engrais.
Les premières réalisations pratiques effectuées dans cette voie ont consisté en synthèses indirectes de l’ammoniac: préparation de cyanures par action, à haute température, de l’azote sur le carbone en présence de bases, puis hydrolyse de ces sels, obtention de cyanamide calcique (CaCN2) qu’on hydrolyse en présence de soude:
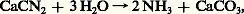
tandis qu’on tentait simultanément de combiner l’azote à l’oxygène afin d’obtenir le monoxyde NO à partir duquel on préparait de l’acide azotique ou des nitrates.
La synthèse directe réalisée industriellement dès 1913 par F. Haber devait supplanter tous ces procédés, sauf la pyrogénation de la houille qui continue à fournir encore un peu d’ammoniac.
Structure
L’ammoniac résulte théoriquement de la mise en commun des trois électrons périphériques 2p de l’azote avec chacun des électrons 1s des trois atomes d’hydrogène. Or, la structure de la molécule montre que celle-ci peut être représentée par une pyramide aplatie, au sommet de laquelle se trouve l’atome d’azote, alors que les trois atomes d’hydrogène sont répartis aux sommets du triangle équilatéral de base. Dans ces conditions, on peut admettre que les cinq électrons 2s et 2p de l’azote donnent lieu par hybridation avec les trois électrons 1s des atomes d’hydrogène à quatre orbitales sp3: trois de celles-ci serviraient à former des liaisons avec les trois atomes d’hydrogène utilisant ainsi six des huit électrons valentiels de la molécule. Le quatrième doublet occuperait la dernière orbitale sp3; le fait que l’angle H 漣 N 漣 H ne mesure que 1070, alors qu’un tel arrangement conduit théoriquement, comme dans le méthane, à un angle tétraédrique de 109,50 s’explique par le fait que, dans le cas de l’ammoniac, les quatre paires d’électrons ne sont pas équivalentes.
Cette molécule dissymétrique a un moment dipolaire de 1,44 debye.
Propriétés physiques
L’ammoniac est un gaz incolore, d’odeur vive, à saveur caustique, irritant les muqueuses. Ses caractéristiques physiques essentielles sont indiquées dans le tableau 1. Elles sont comme celles de l’eau, anormales si on les compare à celles des autres hydrures volatils (phosphine, arsine, stibine). Les températures de fusion et d’ébullition sont plus élevées; sa chaleur d’évaporation anormalement élevée permet, compte tenu de sa basse température d’ébullition, d’utiliser l’ammoniac comme frigorigène.
Ces anomalies s’expliquent par l’existence de liaisons hydrogène entre les atomes d’hydrogène d’une molécule et les atomes d’azote de molécules différentes.
On a élaboré une chimie acidobasique de l’ammoniac liquide calquée sur celle de l’eau.
Grâce à sa constante diélectrique très élevée, l’ammoniac liquide constitue, en effet, comme l’eau, un solvant ionisant pour les électrolytes, compte tenu de sa très faible conductibilité électrique, due à une auto-ionisation peu importante:

caractérisée par une constance de dissociation à 40 0C:
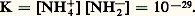
Tous les sels d’ammonium se dissolvent facilement dans l’ammoniac liquide ainsi que la plupart des sels métalliques correspondant à un anion monovalent (nitrates, nitrites, cyanures, halogénures, etc.); au contraire, lorsque l’anion porte plusieurs charges, le sel est généralement insoluble (sulfates, carbonates, phosphates, oxalates, sulfures...). Les oxydes et hydroxydes métalliques sont insolubles.
La comparaison avec l’auto-ionisation de l’eau pure est immédiate et on considérera respectivement, comme acides et comme bases, des produits qui, au sein de l’ammoniac, libéreront respectivement NH4+ et NH2- au lieu de H+ et OH-.
Dans l’ammoniac liquide, les sels d’ammonium possèdent donc une fonction acide; il en est de même de tous les composés se comportant comme des acides en milieu aqueux, car la libération de protons est équivalente à la libération d’ions ammonium par réaction sur le solvant:

les bases sont les amidures métalliques.
La neutralisation d’un acide par une base provoque aussi la formation d’un sel, avec libération d’une ou de plusieurs molécules de solvant:
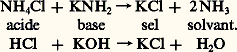
L’équivalence des deux systèmes acide-base est totale. Dans le cas de l’eau, on passe d’un hydroxyde à un oxyde par perte d’une molécule d’eau; dans le cas de l’ammoniac, on passe d’un amidure à un nitrure par perte d’ammoniac:
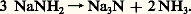
Comme les hydroxydes de zinc et d’aluminium, les amidures correspondants, ainsi que ceux des métaux alcalins et alcalino-terreux, ceux du plomb, de l’étain, du magnésium présentent un caractère amphotère et la base précipitée au cours d’une réaction se redissout dans un excès de réactif:
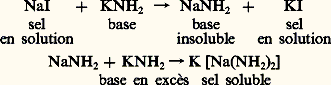
L’action des sels d’ammonium sur les métaux dans l’ammoniac liquide est semblable à celle des acides en phase aqueuse: on obtient un sel du métal attaqué et un dégagement d’hydrogène:

Les métaux dont l’hydroxyde a un caractère amphotère sont attaqués par les bases, également avec dégagement d’hydrogène:

Des réactions plus complexes, comme l’hydrolyse , avec précipitation de sels basiques, trouvent aussi leur équivalent dans l’ammoniac liquide par réaction d’ammoniolyse:

Cela montre bien, comme le pensait le chimiste américain E. C. Franklin dès 1912, que l’eau ne constitue qu’un solvant parmi les autres, et que tout liquide auto-ionisé devait permettre de définir une chimie acido-basique particulière. Ajoutons que l’ammoniac liquide dissout aisément les métaux alcalins et alcalino-terreux et que les solutions ainsi obtenues présentent des propriétés caractéristiques.
Soulignons enfin que l’ammoniac liquide possède une grande affinité pour l’eau et qu’on a pu l’utiliser comme déshydratant basique, notamment pour éliminer l’eau la moins liée des gels de silice et d’alumine, en montrant ainsi que H2Si3 et H2Si25 constituent des individus chimiques.
Le gaz ammoniac se dissout très aisément dans l’eau avec dégagement de chaleur, en donnant de l’ammoniaque.
Sa très grande solubilité dans l’eau (1 176 l.l-1 à 0 0C; 702 l.l-1 à 20 0C) traduit une grande affinité mutuelle de ces deux composés, qui s’explique en partie par leur caractère fortement polaire. L’analyse thermique du système eau-ammoniac montre, d’une part, l’existence de deux composés définis: NH3 練 H2O ( f = 漣 79 0C), et 2 NH3 練 H2O ( f = 漣 78,8 0C), d’autre part, l’absence d’azéotrope; la distillation permet donc de séparer l’ammoniac pur à partir des solutions d’ammoniaque. Ces solutions aqueuses contiennent des liaisons hydrogène N ... H – O; c’est en effet l’eau qui cède ses protons à l’ammoniac, et on est en présence d’hydrate d’ammonium et non d’hydroxyde d’ammonium, lequel n’existe qu’à l’état dissocié NH4+ et OH-; la réaction de dissociation de l’ammoniac dans l’eau s’écrit ainsi:
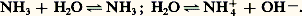
En milieu aqueux, l’ammoniac est donc une base au sens d’Arrhenius, puisqu’il y a libération d’ions hydroxyles. Il s’agit en fait d’une base faible caractérisée par une constante de dissociation très faible (Kb = 1,85 練 10-5, ou pKb = 4,73): dans une solution décimolaire, la dissociation n’est que de l’ordre de 1 p. 100.
La neutralisation par un acide fort ne dégage que 51,5 kJ.mol-1 contre 57,3 dans le cas d’une base forte.
Du point de vue de Brønstedt, NH3 constitue encore une base, puisque par fixation d’un proton il forme l’acide conjugué NH+4:
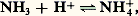
et l’on définit la même constante de dissociation:

mais l’on considère plus fréquemment la constante de dissociation relative à l’équilibre:
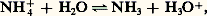
c’est-à-dire l’acide NH4+:

qui est, en fait, relié à Kb par la relation:
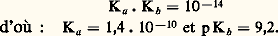
Mais l’existence d’un doublet libre sur l’atome d’azote de la molécule d’ammoniac permet de considérer aussi ce composé comme une base de Lewis: c’est à ce titre qu’il réagira sur le fluorure de bore, lequel constitue un accepteur d’électrons.
Propriétés chimiques
Il convient de distinguer les propriétés du gaz ammoniac de celles de ses solutions aqueuses (ammoniaque). Dans la molécule à l’état gazeux, l’azote est au degré d’oxydation minimal (–3), ce qui explique ses propriétés réductrices. La présence d’un doublet libre, qui traduit une non-saturation de la molécule, se manifeste par des réactions d’addition; enfin certaines réactions de substitution lui confèrent des propriétés acides.
L’ammoniac est relativement peu stable. Il se décompose aisément lorsqu’on le chauffe au contact des métaux:

Ce craquage est mis à profit pour réaliser, lors du traitement thermique des métaux et alliages, des atmosphères conditionnées.
Cette tendance à la décomposition, avec formation d’hydrogène, explique les propriétés réductrices de ce composé qui brûle dans l’oxygène:

En présence d’une toile de platine portée à 700-800 0C l’azote passe au degré + 2 en formant du monoxyde d’azote (oxyde azotique). C’est la réaction de base de la synthèse de l’acide nitrique:

L’oxydation ménagée opérée en présence de méthane donne lieu à la formation d’acide cyanhydrique ; c’est ainsi qu’on réalise industriellement la synthèse de cet acide:

L’ammoniac brûle dans le chlore en donnant de l’acide chlorhydrique et du chlorure d’ammonium; s’il ne s’enflamme pas immédiatement, il peut se produire du trichlorure d’azote NCl3 qui est explosif.
L’ammoniac se fixe sur de nombreux composés en donnant des ammoniacates comparables aux hydrates, mais en général moins stables, c’est-à-dire plus aisément dissociables: 2AgCl 練 3NH3 et AgCl 練 3NH3 notamment. Il réagit également sur les acides pour former les sels d’ammonium qui sont volatils et généralement dissociables:

Avec l’anhydride carbonique, sous pression, il fournit le carbamate d’ammonium:
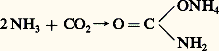
qui par déshydratation se transforme en urée:

Par action de l’ammoniac sur du sodium ou du potassium porté à 400 0C, il se forme un amidure alcalin, blanc, cristallisé, selon la réaction:

qui peut être considérée comme une réaction de substitution. Ces amidures solubles dans l’ammoniac liquide et décomposables par l’eau se présentent comme les sels d’un acide très faible: l’ammoniac.
La formation d’amines R 漣 NH2, par action de l’ammoniac sur les alcools ou sur les halogénures d’alcoyles, et celle d’amides par action sur les chlorures d’acides constituent également des réactions de substitution.
On a indiqué précédemment que l’ammoniac en solution aqueuse ou ammoniaque constituait une base faible. Les sels d’ammonium souvent isomorphes de ceux de potassium sont hydrolysables, volatils et dissociables. C’est à ce titre de base que l’ammoniaque précipite les hydroxydes insolubles de leurs solutions salines:
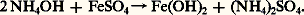
En présence de nombreux sels, l’ammoniaque fournit des ammoniacates et, notamment avec ceux de cobalt, de nickel, de cuivre, d’argent, etc., des complexes qu’on appelle des ammines : par exemple le chlorure d’hexammine cobalt III:

Les solutions d’ammoniac sont oxydables; ainsi, avec le chlore, le brome ou l’hypobromite il se forme de l’azote:

En présence d’hypochlorite on obtient d’abord la chloramine:
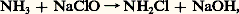
puis l’hydrazine:
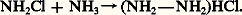
En présence de cuivre, l’ammoniaque fixe l’oxygène de l’air en donnant une solution bleu céleste dite liqueur de Schweizer qui contient de l’hydroxyde de cuivre II tétrammine [Cu(NH3)4](OH)2, base forte qui dissout la cellulose.
Ce caractère oxydable de l’ammoniac explique les transformations que subit ce composé dans le sol, sous l’action de certaines bactéries: les sels ammoniacaux donnent d’abord des nitrites, puis des nitrates, c’est le phénomène de la nitratation démontré par J. J. Schlœsing et son élève A. Muentz et par S. Winogradski.
Production d’ammoniac
L’ammoniac, fixé à l’état de sulfate ou de phosphate d’ammonium lors du traitement des gaz de fours à coke, ne représente qu’un faible pourcentage de l’ammoniac préparé par synthèse directe; celui-ci constitue donc la base de l’industrie des engrais azotés. Il s’agit là d’une fabrication très importante et en pleine expansion; sa production annuelle mondiale était en effet de l’ordre de 100 millions de tonnes au début de la décennie 1990 et, en France comme aux États-Unis, les tonnages d’ammoniac produits, toujours exprimés en tonnes d’azote , ont sensiblement doublé de 1958 à 1965, puis de 1965 à 1981. Depuis lors, la production s’est stabilisée aux États-Unis vers 12 600 000 tonnes d’azote par an, tandis qu’en France, après s’être maintenue fixe, elle a diminué pour atteindre seulement 1 600 000 tonnes d’azote en 1991 (tabl. 2).
Dans les pays gros producteurs (plus d’un million de tonnes par an), on constate depuis 1990 une croissance en Inde (7 022 000 t), aux Pays-Bas (3 162 000 t), en Indonésie (2 790 000 t), au Mexique (2 163 000 t), au Pakistan (1 214 000 t), une stabilité au Japon (1 531 000 t), en Trinité-etTobago (1 520 000 t), en Grande-Bretagne (1 148 000 t), au Canada (2 963 000 t), une décroissance dans l’ex-U.R.S.S. (18 200 000 t), en Allemagne (2 734 000 t), en Pologne (1 608 000 t) et en Italie (1 200 000 t). Toutes les productions qui précèdent entre parenthèses se rapportent à 1990. En 1988, la Chine a produit 16 237 000 tonnes, la Roumanie 3 710 000 tonnes.
La synthèse de l’ammoniac repose sur la réaction équilibrée:

qui est exothermique et s’opère avec diminution de volume: la teneur en ammoniac des gaz à l’équilibre sera donc accrue par une élévation de pression et un abaissement de la température (tabl. 3).
Soulignons que de tels taux de transformation, qui correspondent à l’établissement de l’équilibre, sont très éloignés de ceux que l’on peut prétendre obtenir dans un réacteur industriel, dans lequel les gaz ne séjournent qu’un temps déterminé, appelé durée de contact ; la nécessité d’adopter des températures suffisamment basses pour que le taux de transformation soit encore satisfaisant exige, compte tenu de la lenteur avec laquelle s’opère alors la réaction, l’emploi d’un catalyseur. On est donc amené à travailler sous pression vers 400-500 0C en présence de fer, activé par des promoteurs (alumine et oxyde de potassium).
F. Haber, qui, en collaboration avec C. Bosch, a mis au point au début du XXe siècle la synthèse industrielle, opérait sous 250 bars (1 bar = 105 Pa). Georges Claude, qui a réalisé cette synthèse en France en 1917, a adopté des hyperpressions de 1 000 bars. Par la suite, divers procédés (Fauser, Casale) ont utilisé des pressions intermédiaires de 400 à 600 bars qui, combinées à une température de l’ordre de 400-500 0C, permettent d’atteindre des taux de transformation de l’ordre de 20-25 p. 100; on sépare par liquéfaction l’ammoniac des gaz n’ayant pas réagi et l’on recycle ceux-ci.
La nouvelle tendance consiste à opérer sous des pressions plus faibles et les six nouvelles unités de 1 000 tonnes par jour qui ont été construites en France entre 1965 et 1990, travaillent sous des pressions qui ne dépassent pas 150 à 250 bars.
Si les modes de préparation du mélange gazeux peuvent être assez différents d’une usine à l’autre, même lorsqu’elles utilisent la même matière première (le gaz naturel, par exemple), les ateliers de synthèse sont beaucoup plus comparables entre eux; ils comportent tous une boucle de synthèse dont le fonctionnement est indiqué sur la figure, qui schématise la boucle d’un atelier Fauser. Un mélange gazeux purifié et comprimé (A) est mélangé en B aux gaz sortant du tube de synthèse. Le gaz résultant, déshuilé, traverse le réfrigérant-condenseur R. L’ammoniac liquide, qui se sépare en S, dissout les faibles quantités d’argon introduit avec l’azote, et le méthane provenant de l’hydrogénation de traces de monoxyde de carbone présent dans le mélange gazeux. Les gaz saturés d’ammoniac passent dans l’échangeur E où ils s’échauffent en refroidissant les gaz issus du tube T; puis, après avoir été portés à la pression convenable par le compresseur C, ils sont introduits dans le tube T où ils sont portés à la température voulue, aux dépens des gaz ayant traversé le catalyseur. Celui-ci est disposé à la partie supérieure du tube, qui constitue le réacteur proprement dit. Après avoir été mélangés aux gaz frais en B, les gaz sont refroidis dans le réfrigérant R. Éventuellement, une purge P permet de limiter la proportion de méthane et d’argon qui risquent de s’accumuler dans le gaz, malgré leur solubilité dans l’ammoniac liquide.
Les réacteurs, dont la mise au point est délicate, sont construits classiquement en acier sans carbone et sont appliqués à d’autres synthèses, comme celle du méthanol. La chambre de catalyse occupe généralement la partie supérieure du tube, tandis que la partie inférieure constitue un échangeur thermique: la réaction étant exothermique, on s’est en effet évertué à porter les gaz à la température de la réaction (400-500 0C) sans source extérieure de chaleur. Par la suite, G. Fauser a réussi à utiliser la chaleur dégagée, non seulement pour préchauffer les gaz, mais pour produire de la vapeur (de l’ordre de 0,9 t par tonne d’ammoniac) en disposant dans la chambre de catalyse des circuits (dits épingles) traversés par de l’eau qui se vaporise. On parvient ainsi à établir dans le lit du catalyseur un profil de températures tel que, dans chaque tranche, la température est adaptée à la composition du mélange qui la traverse, améliorant ainsi le taux de transformation des gaz.
L’atelier qui vient d’être décrit est dit autonome car il fonctionne sans apport d’énergie extérieure: il utilise l’effet thermique de la réaction pour porter les gaz introduits à la température convenable et produit de la vapeur en quantité suffisante pour fournir l’électricité nécessaire aux pompes, etc.
Les usines de 1 000 t/j sont également autonomes bien que le réacteur ne comporte pas d’«épingles» à circulation de vapeur, comme dans le tube de synthèse Fauser. La vapeur est produite par circulation des gaz, issus du réacteur T, dans une chaudière placée entre celui-ci et l’échangeur. Toutefois les épingles avaient également un autre objet: elles étaient disposées de telle sorte que des débits d’eau convenables portaient les diverses couches du catalyseur à des températures telles qu’on réalisait le profil idéal de température. Il convient en effet de souligner que la température optimale de réaction dépend de la composition du mélange gazeux et que celle-ci varie au fur et à mesure de sa transformation en ammoniac.
On obtient plus aisément le même résultat en substituant aux épingles des injecteurs par lesquels sont admis dans le réacteur des débits gazeux déterminés du mélange issu de l’échangeur E: on provoque ainsi des trempes locales du gaz dans le réacteur.
La technique du procédé de synthèse n’a subi qu’une évolution normale, tendant à accroître la capacité de production des ateliers. Celle-ci atteint et dépasse même 1 000 t/j grâce à l’emploi de compresseurs centrifuges. La préparation du mélange réactionnel a par contre connu une véritable révolution quant aux matières premières utilisées comme source d’hydrogène, l’azote étant toujours extrait de l’air.
Jusqu’en 1940, on peut dire que, mis à part quelques ateliers de synthèse, préparant l’hydrogène par électrolyse de l’eau, ou utilisant l’hydrogène résiduaire obtenu lors de la fabrication du chlore par électrolyse des solutions de chlorure de sodium, la fabrication de l’hydrogène s’opérait à partir de la houille.
Après la Seconde Guerre mondiale, les produits pétroliers se sont substitués à la houille comme matière première de l’ammoniac. Ainsi, aux États-Unis, 90 p. 100 de l’ammoniac seraient obtenus à partir de produits pétroliers.
L’extraction de l’hydrogène des gaz de fours à coke, qui en contiennent de l’ordre de 50 p. 100, s’opérait par liquéfaction et distillation fractionnée.
On séparait simultanément une fraction riche en méthane (le gaz de four à coke en contient environ 25 p. 100) qu’on traitait généralement, pour en extraire de l’hydrogène, par une technique comparable à celle appliquée au gaz naturel. La préparation de l’hydrogène à partir de ce dernier, constitué essentiellement de méthane, a donné lieu à de très nombreux procédés industriels car elle peut s’opérer:
– par simple craquage avec formation simultanée de noir de carbone:

– par conversion à la vapeur d’eau:

– soit enfin par conversion à l’oxygène:

qui, en fait, donne lieu à des réactions plus complexes.
Le craquage simple qui exige de travailler à haute température n’est plus utilisé; par contre, diverses usines pratiquent la conversion à la vapeur d’eau, soit en discontinu et sans catalyseur (sur un garnissage porté à 1 200 0C par combustion du méthane), soit en présence de catalyseur, soit encore en continu à 800 0C avec un chauffage externe.
La conversion par l’oxygène, qui fait l’objet d’un procédé O.N.I.A. (Office national industriel de l’azote), est appliquée dans plusieurs usines françaises; elle s’opère généralement à 800 0C sur un catalyseur au nickel. On obtient un mélange d’oxyde de carbone et d’hydrogène qu’on soumet à la «conversion». Par action sur la vapeur d’eau à 500 0C en présence de catalyseurs (oxydes de fer et d’alumine), l’oxyde de carbone fournit de l’hydrogène et de l’anhydride carbonique, qu’on sépare par dissolution dans l’eau sous pression, ou par lavage avec des solutions de soude ou d’éthanolamines.
On substitue parfois à l’oxygène de l’air suroxygéné; l’azote nécessaire à l’obtention du mélange stoechiométrique, obtenu précédemment par liquéfaction et distillation de l’air, est de plus en plus introduit en même temps que l’oxygène nécessaire à la conversion du méthane, de telle sorte que les ateliers peuvent alors se dispenser d’une installation de liquéfaction d’air.
Soulignons que le mélange réactionnel doit être soumis, au cours de sa compression qui s’opère en plusieurs stades, à une purification poussée, ayant pour objet d’éliminer les traces de C2 (potasse) et de CO (lessives cuivreuses) présents dans le gaz (CO2 et CO se transformeraient en méthane et CO empoisonnerait le catalyseur) et de dessécher le gaz avant de l’envoyer à l’atelier de catalyse.
Utilisations
De 80 à 90 p. 100 de l’ammoniac produit sont utilisés comme engrais, soit directement sous forme de sels d’ammonium, sulfate, phosphate, soit après transformation à l’état de sels de l’acide nitrique (nitrate d’ammonium, nitrate de potassium) ou d’urée.
On a mis au point aux États-Unis l’utilisation non seulement d’engrais liquides constitués par des solutions aqueuses de composés «azotés»: nitrate d’ammonium, urée additionnée ou non d’ammoniac (par exemple 60 p. 100 d’urée et 26 p. 100 d’ammoniac), mais aussi d’ammoniac liquide; il est alors enfoui à 10 cm dans le sol où il se volatilise et se fixe sur l’humus; c’est la nitrojection. Ce procédé a été expérimenté dans certaines régions de France, mais il ne semble pas devoir s’étendre; par contre, l’utilisation d’engrais liquides préparés dans des centres agricoles est en plein développement.
L’ammoniac est utilisé aussi comme agent de nettoiement, pour la fabrication des pâtes à papier, et en métallurgie pour réaliser les traitements thermiques. Il constitue également la matière première des synthèses de l’acide nitrique et du nitrate d’ammonium (explosif), de l’acide cyanhydrique et de l’urée (obtention de matières plastiques: aminoplastes), de l’acrylonitrile, des amines, et du perchlorate d’ammonium (propergol pour fusées).
En thérapeutique, l’ammoniac est employé en solutions diluées, soit en usage externe (liniments, composés rubéfiants ou révulsifs, solutions contre les piqûres d’insectes), soit en inhalations, comme excitant de la respiration et de la circulation (dans les syncopes).
Chez l’homme, après dégradation des protéines, l’ammoniac est fixé par les acides glutamiques et aspartiques. On le retrouve dans l’urine sous forme d’ions ammonium (NH4+)et surtout sous forme d’urée.
Toxicologie
L’ammoniac est un gaz toxique, il irrite les muqueuses (surtout oculaires). Une atmosphère à 1 p. 100 serait mortelle en 5 minutes. L’ammoniaque a des propriétés vésicantes et caustiques. Absorbée par la bouche, en solution concentrée et en dose assez importante, elle produit une irritation de l’appareil digestif. Le malade souffre de vives douleurs accompagnées de diarrhées et de vomissements.
ammoniac, iaque [ amɔnjak ] adj. et n. m.
• armoniac XIVe; lat. ammoniacum « de la région du temple d'Ammon », en Libye
1 ♦ Gomme ammoniaque : gomme-résine d'une plante d'Afrique. Vx Sel ammoniac : chlorure d'ammonium.
2 ♦ (1787) Gaz ammoniac, ou n. m. ammoniac : combinaison gazeuse d'azote et d'hydrogène (NH3), gaz à odeur piquante, facilement liquéfiable, issu à l'état naturel de la décomposition des matières organiques azotées, préparé industriellement par synthèse, utilisé en particulier pour la préparation des sels ammoniacaux (engrais) et de l'ammoniaque.
⊗ HOM. Ammoniaque.
● ammoniac nom masculin (latin ammoniacum, du grec Ammoniakon, de Ammôn, nom grec d'Amon, dieu égyptien, parce qu'on préparait autrefois cette substance en Libye, près de son temple) Combinaison gazeuse d'azote et d'hydrogène de formule NH3 existant à l'état libre ou dissoute dans l'eau. (Cette solution, qui ne constitue pas une combinaison, est appelée ammoniaque.) ● ammoniac (difficultés) nom masculin (latin ammoniacum, du grec Ammoniakon, de Ammôn, nom grec d'Amon, dieu égyptien, parce qu'on préparait autrefois cette substance en Libye, près de son temple) Orthographe Avec deux m. Sens Ne pas confondre ces deux homonymes. Ammoniac, avec un c, pour le composé gazeux. Ammoniaque, avec -que, pour la solution aqueuse. ● ammoniac (expressions) nom masculin (latin ammoniacum, du grec Ammoniakon, de Ammôn, nom grec d'Amon, dieu égyptien, parce qu'on préparait autrefois cette substance en Libye, près de son temple) Sel ammoniac, nom commercial du chlorure d'ammonium. ● ammoniac (homonymes) nom masculin (latin ammoniacum, du grec Ammoniakon, de Ammôn, nom grec d'Amon, dieu égyptien, parce qu'on préparait autrefois cette substance en Libye, près de son temple) ammoniaque nom féminin
ammoniac, aque
adj. Sel ammoniac: chlorure d'ammonium.
————————
ammoniac
n. m. Gaz composé d'azote et d'hydrogène (NH 3), incolore et d'odeur suffocante, extrêmement soluble dans l'eau, utilisé notam. pour la fabrication d'engrais.
⇒AMMONIAC, AQUE, subst. et adj.
CHIMIE
I.— Ammoniac, subst. masc. et adj.
A.— Subst. masc. Gaz incolore, possédant une odeur forte caractéristique, suffocant, qui se dégage lors de la décomposition de matières organiques azotées sous l'influence de certains ferments. Synon. gaz ammoniac :
• 1. Haber réussit à appliquer les principes de l'équilibre à la réaction de synthèse de l'ammoniac à partir d'azote et d'hydrogène.
Hist. générale des sciences, t. 3, vol. 2, 1964, p. 406.
Rem. 1. Certains auteurs confondent l'ammoniac qui est un gaz et l'ammoniaque (cf. infra) qui est la solution aqueuse de ce gaz. Ainsi s'agit-il de l'ammoniaque (liquide) dans l'ex. suiv. :
• 2. ... on verse de l'ammoniac goutte à goutte avec une pipette...
NICOLAS ds (F. Widal, P.-J. Teissier, G.-H. Roger, Nouveau traité de médecine, fasc. 4, 1920-1924, p. 591).
Rem. 2. Inversement ammoniaque a été parfois employé au sens d'ammoniac :
• 3. L'ammoniaque est un gaz incolore ...
DESCHAMPS D'AVALLON, Compendium de pharmacie pratique, 1868, p. 799.
B.— Emploi adj. :
• 4. Le gaz ammoniac (...) est très soluble dans l'eau ...
P. LEBEAU, G. COURTOIS, Traité de pharmacie chimique, t. 1, 1929, p. 188.
♦ Spéc., vx. Sel ammoniac. Substance blanche résultant de la combinaison de l'acide chlorhydrique et de l'ammoniac. Synon. mod. chlorure d'ammonium :
• 5. J'essayai de former de l'alcali volatil en décomposant le sel ammoniac avec de l'alcali fixe; mais la production fut lente et peu sensible, tandis qu'au niveau de la mer, cette production, par la même dose, me parut prompte et très-abondante.
Voyage de La Pérouse autour du monde, t. 4, 1797, p. 3.
II.— Ammoniaque, subst. fém.
A.— Solution obtenue par dissolution du gaz ammoniac dans l'eau. Synon. vx, esprit de sel ammoniac, alcali volatil :
• 6. D'ailleurs, les plaies qu'ils [les moustiques] causent sont rapidement abolies par une goutte d'ammoniaque diluée dans l'eau.
H. COUPIN, Animaux de nos pays, dict. pratique, 1909, p. 309.
• 7. Pour aller chez Vévé, on franchissait d'abord un vaste porche commercial, qui sentait l'ammoniaque, les chevaux et la buanderie. Une maison de produits pharmaceutiques encombrait toute l'entrée.
J. MALÈGUE, Augustin ou le Maître est là, t. 1, 1933, p. 278.
• 8. Autour de notre planète naissante, en plus des premières ébauches d'une barysphère métallique, d'une lithosphère silicatée, d'une hydrosphère et d'une atmosphère, il y a donc lieu de considérer les linéaments d'une enveloppe spéciale — antithèse, pourrait-on dire, des quatre premières : zone tempérée de la polymérisation, où eau, ammoniaque, acide carbonique, flottent déjà, baignée de rayons solaires. Négliger cette buée ténue serait priver l'astre juvénile de sa plus essentielle parure.
P. TEILHARD DE CHARDIN, Le Phénomène humain, 1955, p. 70.
Rem. Cette solution est employée pour l'usage domestique comme dégraissant :
• 9. Il empoigna résolument les flacons de parfums, débarbouilla les goulots et les bouchons à l'émeri, frotta les étiquettes avec de la gomme élastique et de la mie de pain, puis il savonna la cuvette, trempa les peignes et les brosses dans de l'eau saturée d'ammoniaque, fit manœuvrer son vaporisateur et injecta la pièce de poudre de lilas de Perse, lava les toiles cirées du parquet et du mur, étrilla le petit cheval, essuya le dossier et les barreaux de la chaise basse.
J.-K. HUYSMANS, Là-bas, t. 1, 1891, p. 238.
— Par métaph. :
• 10. Ainsi, l'image de Marina, évoquée dans l'atmosphère allemande, l'illuminait pour André. Alors qu'il s'épaississait avec ces socialistes qui préparent le bonheur futur comme un festin de noce, le souvenir de Marina fut une goutte d'ammoniaque versée dans son verre à la suite d'un repas trop lourd, et qui restitue à l'esprit sa lucidité.
M. BARRÈS, L'Ennemi des Lois, 1893, p. 186.
— Emploi adj. PHARM., vx. Gomme-résine d'odeur fétide tirée d'une plante africaine, la dorème : Gomme ammoniaque :
DORVAULT, L'Officine, 1844, p. 150.
B.— Vx. Sel résultant de la combinaison de l'ammoniaque avec un acide. Synon. ammonium :
• 12. La fabrication du chlorure d'ammonium, appelé aussi chlorhydrate d'ammoniaque, etc.
P. LEBEAU, G. COURTOIS, Traité de pharmacie chimique, t. 1, 1929, p. 43.
• 13. Le phosphate d'ammoniaque fut le premier en date de ces engrais complexes. Ses progrès résultèrent directement de ceux de l'industrie du phosphore. La première usine de phosphate d'ammoniaque fut créée en 1838 par Coignet, à Lyon, mais c'est seulement au début du XXe siècle qu'il commença de devenir un produit courant.
L'Industrie française des engrais chimiques, 1. Engrais phosphatés et engrais azotés, 1954, p. 12.
DÉR. Ammoniacé, ée, ammonacé, ée, adj., chim. Qui contient de l'ammoniaque (attesté ds NYSTEN 1814 et la plupart des dict. gén. du XIXe s.). Ammoniate, subst. masc., chim., synon. de ammoniure (attesté ds NYSTEN 1814, Ac. Compl. 1842, BESCH. 1845, Lar. 19e). Ammonimétrie, subst. fém., chim. ,,Dosage de l'ammoniaque`` (HOUZEAU, Ac. des sc. Comptes rend., t. 84, 1877, p. 551 ds LITTRÉ). Ammonique, adj., chim., synon. de ammoniacal (attesté ds Ac. Compl. 1842 et la plupart des dict. des XIXe et XXe s.). Ammonisation, subst. fém., chim. Formation de sels ammoniacaux (attesté ds Lar. 20e et PLANTEFOL, Cours de botanique et de biologie végétale, t. 1, 1931, p. 359). Ammoniure, subst. masc., chim. Nom donné aux composés résultant de la combinaison de l'ammoniaque avec certains oxydes métalliques; ammoniure de cuivre (cf. MOZIN-BIBER, t. 1 1811 et DESCHAMPS D'AVALLON, Compendium de pharmacie pratique, 1868, p. 813).
Prononc. ET ORTH. — 1. Forme phon. :[ ]. PASSY 1914, BARBEAU-RODHE 1930, Pt ROB. et Pt Lar. 1968 transcrivent le mot avec un seul m. WARN. 1968 signale également la possibilité d'une prononc. avec m géminé (cf. aussi LAND. 1834, GATTEL 1841, LITTRÉ et DG pour m géminé). 2. Forme graph. — À propos de la graph. ammoniac, ammoniaque, GREV. 1964, § 345 fait remarquer : ,,Quelques adjectifs terminés par -c : ammoniac, caduc, franc (peuple), public, turc, remplacent c par qu devant l'e du féminin : Ammoniaque, caduque, franque, publique, turque``. FÉR. 1768 écrit : ammoniac ou armoniak. — Dér. Ammoniate. Seule transcription ds LITTRÉ : a-mmo-ni-a-t'. Ammonimétrie. Seule transcription ds LITTRÉ : a-mmo-ni-mé-trie. Ammonisation : [
]. PASSY 1914, BARBEAU-RODHE 1930, Pt ROB. et Pt Lar. 1968 transcrivent le mot avec un seul m. WARN. 1968 signale également la possibilité d'une prononc. avec m géminé (cf. aussi LAND. 1834, GATTEL 1841, LITTRÉ et DG pour m géminé). 2. Forme graph. — À propos de la graph. ammoniac, ammoniaque, GREV. 1964, § 345 fait remarquer : ,,Quelques adjectifs terminés par -c : ammoniac, caduc, franc (peuple), public, turc, remplacent c par qu devant l'e du féminin : Ammoniaque, caduque, franque, publique, turque``. FÉR. 1768 écrit : ammoniac ou armoniak. — Dér. Ammoniate. Seule transcription ds LITTRÉ : a-mmo-ni-a-t'. Ammonimétrie. Seule transcription ds LITTRÉ : a-mmo-ni-mé-trie. Ammonisation : [ ] (cf. Pt ROB.). Ammoniure. Seule transcription ds LITTRÉ : a-mmo-ni-ur'.
] (cf. Pt ROB.). Ammoniure. Seule transcription ds LITTRÉ : a-mmo-ni-ur'.
]. PASSY 1914, BARBEAU-RODHE 1930, Pt ROB. et Pt Lar. 1968 transcrivent le mot avec un seul m. WARN. 1968 signale également la possibilité d'une prononc. avec m géminé (cf. aussi LAND. 1834, GATTEL 1841, LITTRÉ et DG pour m géminé). 2. Forme graph. — À propos de la graph. ammoniac, ammoniaque, GREV. 1964, § 345 fait remarquer : ,,Quelques adjectifs terminés par -c : ammoniac, caduc, franc (peuple), public, turc, remplacent c par qu devant l'e du féminin : Ammoniaque, caduque, franque, publique, turque``. FÉR. 1768 écrit : ammoniac ou armoniak. — Dér. Ammoniate. Seule transcription ds LITTRÉ : a-mmo-ni-a-t'. Ammonimétrie. Seule transcription ds LITTRÉ : a-mmo-ni-mé-trie. Ammonisation : [] (cf. Pt ROB.). Ammoniure. Seule transcription ds LITTRÉ : a-mmo-ni-ur'.Étymol. ET HIST.
I.— Adj. XIVe s. chim. sal armoniac « chlorure d'ammonium » (BRUN DE LONG BORC, f° 72d ds GDF. Compl.), vedette maintenue jusqu'à Trév. 1752; 1575 gomme ammoniac « gomme-résine d'une plante d'Afrique » (PARÉ, V, 20, ibid.).
II.— Subst. 1359 armonial « chlorure d'ammonium » (Compte de l'argent, p. 236 ds GDF. Compl.); 1782 ammoniac (GUYTON DE MORVEAU, Mémoire sur les démonstrations chimiques, la nécessité d'en perfectionner le système, Dijon ds Journal d'observations sur la physique ..., p. 379 : Le mot ammoniac n'a eu seul et par lui-même jusqu'à présent aucune signification; prenons-le pour le nom de l'être simple qui nous manque); 1787 ammoniaque (ID., Méthode d'une nomenclature chimique, tableau p. 100 : Substance non décomposée. Nom nouveau : L'Ammoniaque).
Empr. au lat. ammoniacum, adj. sal ammoniacus (PLINE, 31, Hist. nat., 7, 39, 78 ds FORC. 1864-1926 : Quod vero in officinis est sale ammoniaco vel armoniaco, facticius est, vel saltem ab illo adulteratus); subst. ammoniacum « gomme résine » (PLINE, 12, Hist. nat., 23, 49, 107, ibid.); gr. 
 « sel ammoniac ou gomme ammoniaque » (DIOSCORIDE, 3, 98 ds BAILLY), dér. de
« sel ammoniac ou gomme ammoniaque » (DIOSCORIDE, 3, 98 ds BAILLY), dér. de  parce qu'on recueillait ces produits près du temple de Zeus Ammon en Libye; ammoniaque, fém. substantivé de ammoniac.
parce qu'on recueillait ces produits près du temple de Zeus Ammon en Libye; ammoniaque, fém. substantivé de ammoniac.
« sel ammoniac ou gomme ammoniaque » (DIOSCORIDE, 3, 98 ds BAILLY), dér. de parce qu'on recueillait ces produits près du temple de Zeus Ammon en Libye; ammoniaque, fém. substantivé de ammoniac.— Ammoniacé, ammoniate, 1814-1820 (NYSTEN). Ammonimétrie 1877, supra rem. Ammonique, 1838 (Ac. Compl. 1842). Ammonisation, 1928 (Lar. 20e). Ammoniure, 1811 (MOZIN-BIBER t. 1).
STAT. — Fréq. abs. litt. :3.
BBG. — BADER-TH. 1962. — BÉL. 1957 (et s.v. ammoniacé, ée, ammoniate, ammoniure). — BOISS.8. — BOUILLET 1859 (et s.v. ammoniure). — BRARD 1838. — CHESN. 1857 (et s.v. ammoniacé, ammonique, ammoniure). — Comm. t. 1 1837. — DELORME 1962. — DUVAL 1959 (et s.v. ammonacé, ammoniacé, ammoniate, ammonique, ammonisation, ammoniure). — FÉR. 1768. — FROMH.-KING. 1968 (et s.v. ammoniate). — GALIANA Astronaut. 1963. — GALIANA Déc. sc. 1968. — GRAND. 1962. — HANSE 1949. — Lar. comm. 1930. — Lar. méd. 1970. — Lar. mén. 1926. — LITTRÉ-ROBIN 1865 (et s.v. ammoniacé, ée, ammoniate, ammonique, ammoniure). — Méd. Biol. t. 1 1970 (s.v. ammoniacé, ammonique). — MONT. 1967. — NYSTEN 1824 (et s.v. ammoniacé, ammoniate). — PLAIS.-CAILL. 1958 (s.v. ammonisation). — POPE 1961 [1952], § 397. — PRÉV. 1755. — PRIVAT-FOC. 1870. — Sc. 1962. — THOMAS 1956. — UV.-CHAPMAN 1956.
ammoniac [amɔnjak] adj. et n. m.
ÉTYM. 1575, gomme ammoniac; sal armoniac, XIVe, « chlorure d'ammonium », dit aussi armonial (1359); lat. ammoniacum, adj. (Pline), grec ammoniakon, cette gomme étant recueillie en Libye, non loin du temple de Zeus Ammon.
❖
1 Vx. || Sel ammoniac : chlorure d'ammonium.
♦ Gomme ammoniac ou ammoniaque : résine produite par le dorème, plante orientale, d'odeur forte et piquante.
2 Adj. et n. m. (1787, Guyton de Morveau). Chim. et techn. || Gaz ammoniac, ou, n. m., l'ammoniac : combinaison gazeuse d'azote et d'hydrogène (NH3), gaz à odeur piquante, très soluble, facilement liquéfiable, issu à l'état naturel de la décomposition des matières organiques azotées (eaux ammoniacales des cokeries et usines à gaz), de la fermentation des eaux de vidange et de la décomposition de la cyanamide. || Préparé industriellement par synthèse, l'ammoniac est utilisé en particulier pour la préparation des sels ammoniacaux (engrais : chlorhydrate, sulfate, nitrate, phosphate d'ammoniac), de l'ammoniaque, de la soude.
♦ (Anglic.). ⇒ Crude ammoniac.
❖
DÉR. Ammoniacal, ammoniaque, ammoniation, ammoniure, ammoniurie. — V. Amide, amine, ammonisation.
COMP. V. Ammoniaco-, ammonitrate.
HOM. Ammoniaque.
Encyclopédie Universelle. 2012.