* * *
1. [MIM*230400] An inborn error of galactose metabolism due to congenital deficiency of the enzyme galactosyl-1-phosphate uridylyltransferase, resulting in tissue accumulation of galactose 1-phosphate; manifested by nutritional failure, hepatosplenomegaly with cirrhosis, cataracts, mental retardation, galactosuria, aminoaciduria, and albuminuria that regress or disappear if galactose is removed from the diet; autosomal recessive inheritance; caused by mutation in the galactose-1-phosphate uridyltransferase gene (GALT) on 9p. SEE ALSO: galactokinase deficiency. 2. An inborn error in metabolism other than a deficiency in galactosyl-1-phosphate uridylyltransferase (see subentries below). SYN: galactose diabetes. [galactose + G. haima, blood]
- epimerase deficiency g. an inborn error in metabolism in which there is a deficiency of uridine diphosphate galactose 4-epimerase; galactose 1-phosphate accumulates.
- galactokinase deficiency g. an autosomal recessive disorder resulting in an accumulation of galactose and galactitol.
- transferase deficiency g. an autosomal recessive disorder in which there is a deficiency of galactose-1-phosphate uridylyltransferase (see main entry for g.).
* * *
ga·lac·tos·emia or chiefly Brit ga·lac·tos·aemia gə-.lak-tə-'sē-mē-ə n a metabolic disorder inherited as an autosomal recessive trait in which galactose accumulates in the blood due to deficiency of an enzyme catalyzing its conversion to glucose
ga·lac·tos·emic or chiefly Brit ga·lac·tos·aemic -mik adj
* * *
ga·lac·tos·e·mia (gə-lak″to-seґme-ə) [galactose + -emia] a general term encompassing three autosomal recessive disorders resulting from defective galactose metabolism. Classic galactosemia, which is often fatal to neonates, is caused by mutations in the GALT gene (locus: 9p13), which encodes UDP-glucose–hexose-1-phosphate uridylyltransferase. Enzyme deficiency results in accumulation of galactose 1-phosphate and galactose, with cataracts, cirrhosis, hepatomegaly, vomiting, diarrhea, jaundice, poor weight gain, and malnutrition in infancy, and mental retardation in survivors. The other two disorders are galactokinase deficiency and UDP-glucose 4-epimerase deficiency (qq.v.).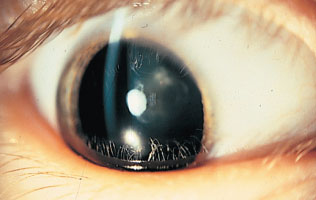
 Cataract associated with galactosemia, the opacity of the lens having an oil-droplet appearance.
Cataract associated with galactosemia, the opacity of the lens having an oil-droplet appearance.
Medical dictionary. 2011.