CORROSION
La corrosion est le résultat de l’action qu’exerce un réactif liquide ou gazeux sur un métal ou un alliage. Sa forme la plus connue est la rouille du fer exposé à l’air humide. Elle présente une grande importance étant donné les conséquences qu’elle a dans des domaines très variés de l’activité humaine. Dans le domaine économique, par exemple, les dommages causés par la corrosion ont une incidence non négligeable sur l’établissement des prix de revient. C’est ainsi que l’on estime à 20 p. 100 environ de la production la quantité d’acier qui est chaque année utilisée pour le remplacement des installations de toutes sortes détruites par la corrosion. Le remplacement du matériel corrodé constitue donc, pour l’industrie en particulier, une charge financière très élevée à laquelle il faut ajouter le manque à gagner correspondant à l’arrêt des installations, nécessaire pour effectuer les réparations.
Les technologies avancées constituent également un bon exemple du rôle fondamental que joue la tenue des matériaux métalliques aux agents corrosifs. En effet, bien souvent, le développement d’une technique est freiné parce qu’il est difficile de trouver un métal ou un alliage qui résiste aux conditions d’emploi toujours plus difficiles qui sont exigées.
Les recherches sur la corrosion sont anciennes puisque, dès 1830, le physicien Auguste De La Rive en proposait une théorie électrochimique. Cependant, ces recherches n’ont véritablement pris leur essor qu’au XXe siècle. Leur but est double: déterminer le processus des phénomènes afin de leur trouver un remède, et définir les matériaux susceptibles d’être utilisés dans des conditions données pendant une durée qui est parfois de plusieurs décennies, comme c’est le cas pour certaines installations nucléaires.
L’étude de la corrosion constitue une branche de la chimie puisqu’elle correspond à des réactions qui font intervenir un métal et un réactif. Mais, pour résoudre les problèmes qu’elle pose, on ne peut pas se contenter d’appliquer les lois classiques de la chimie. En effet, de nombreux facteurs spécifiques, se rapportant au métal ou au réactif, peuvent avoir une grande influence sur la genèse et le développement des phénomènes de corrosion, si bien qu’il est indispensable pour les étudier de faire également appel aux lois de la métallurgie et de l’électrochimie.
1. La corrosion en solution
Les phénomènes de corrosion peuvent se développer suivant différents processus: corrosions chimique, électrochimique, biologique et corrosion accompagnée d’érosion.
La corrosion chimique est le résultat d’une réaction hétérogène entre une phase solide, le métal, et une phase gazeuse ou liquide. Il est très difficile de donner un exemple de corrosion chimique pure par un liquide puisque celle-ci est le plus souvent accompagnée d’une corrosion électrochimique. Cette dernière se manifeste lorsqu’il existe une hétérogénéité soit dans le métal, soit dans le réactif. L’existence de ces hétérogénéités détermine la formation de piles, et un courant électrique circule entre anodes et cathodes, les zones constituant les anodes étant attaquées.
La corrosion biologique correspond à l’attaque directe ou indirecte des métaux par des bactéries. Celles-ci peuvent produire des composés, par exemple le dioxyde de carbone, l’anhydride sulfureux ou des acides organiques qui attaquent le métal. Certaines bactéries comme Desulfovibrio desulfuricans réduisent les sulfates en soufre, et le sulfure de fer peut se former. L’attaque bactérienne apparaît en particulier dans les canalisations enterrées.
La présence de produits de corrosion adhérents et continus à la surface des métaux a souvent pour effet de ralentir le processus de corrosion. Si cette couche est éliminée en certains points par le mouvement du milieu environnant ou par les particules solides qu’il contient, il y a accélération de la corrosion; on dit qu’il y a corrosion par érosion.
En général, ces différents types de corrosion n’interviennent pas indépendamment les uns des autres, ce qui a pour effet de rendre plus délicate l’interprétation des phénomènes.
Morphologie de la corrosion
La corrosion peut se manifester de quatre façons principales. Si elle se développe à la même vitesse en tous les points du métal, il s’agit de corrosion uniforme. C’est le cas de l’attaque du fer par l’eau avec formation de rouille.
Dans certains cas, l’attaque est localisée aux joints de grains du métal. On dit qu’il y a corrosion intergranulaire ou intercristalline (fig. 1). C’est un type de corrosion très dangereux car il se produit en profondeur et, bien que la perte de métal soit faible, il conduit rapidement à une chute des caractéristiques mécaniques du métal. On l’observe dans de nombreux alliages dans lesquels s’est développée une précipitation aux joints de grains.
La corrosion par piqûres se localise en certains points du métal. Elle peut avoir plusieurs origines, en particulier l’existence d’une discontinuité dans la couche protectrice recouvrant le métal (fig. 2).
Enfin, dans certains cas, la corrosion peut se manifester à l’intérieur des cristaux. Il s’agit alors de corrosion intra- ou transcristalline. Elle apparaît souvent dans les phénomènes de corrosion sous contrainte.
Facteurs de la corrosion
Comme toute réaction chimique, la corrosion des métaux dépend des facteurs qui caractérisent le réactif, en particulier de la concentration, du pH et de la température. Les facteurs métalliques y jouent également un rôle fondamental. C’est ainsi que la connaissance de la composition chimique d’un alliage est tout à fait insuffisante pour définir son comportement dans un milieu donné. Trois caractéristiques supplémentaires doivent être connues. Ce sont celles qui résultent des traitements thermiques, mécaniques et superficiels subis par l’alliage. Prenons l’exemple de l’acier inoxydable du type 18-8 (18 p. 100 de chrome, 8 p. 100 de nickel). S’il a subi une hypertrempe depuis 1 150 0C, il résiste très bien à l’attaque par l’acide sulfurique contenant du sulfate de cuivre. En revanche, si à partir de cette même température il est refroidi lentement, il devient très sensible à la corrosion intergranulaire. En effet, au cours du refroidissement, le carbure de chrome Cr23C6 précipite aux joints de grains et sa présence, en provoquant la formation de zones appauvries en chrome, a pour effet de favoriser une corrosion intergranulaire suivant un processus électrochimique.
Les traitements mécaniques déterminent en général un écrouissage du métal, dont la réactivité augmente alors. C’est ainsi que, dans les pièces préparées par emboutissage, la corrosion se manifeste de façon préférentielle dans les régions le plus déformées. C’est le cas de l’attaque des douilles en laiton dans les vapeurs ammoniacales. Un traitement de détente par chauffage de la pièce à 250 0C suffit, en éliminant les tensions, à supprimer le phénomène.
La corrosion se manifestant à l’origine à la surface du métal, il est logique que les caractéristiques de cette surface jouent un rôle important sur le comportement du métal à l’égard de la corrosion. Les caractéristiques superficielles des métaux et alliages qui définissent ce que l’on appelle l’état de surface peuvent être de nature cristallographique, chimique, physico-chimique ou microgéométrique; les caractéristiques cristallographiques dépendent du type de structure, de la dimension des grains et du degré de perfection du réseau cristallin, en relation avec le mode d’élaboration du métal ou de l’alliage et avec ses conditions d’emploi; les caractéristiques chimiques font intervenir aussi le degré de pureté du métal ou de l’alliage, donc la présence d’impuretés, de précipités ou de différentes phases intermétalliques; les caractéristiques physico-chimiques dépendent de la nature du film le plus souvent présent à la surface du métal (films d’oxydes, composés organiques ou gaz adsorbés, qui établissent une barrière entre le métal et le réactif); enfin, les caractéristiques microgéométriques sont définies par le profil du métal, qui détermine en particulier la surface réelle soumise à l’action du milieu extérieur. Elles varient avec les méthodes utilisées pour la mise en forme des matériaux métalliques.
Corrosion électrochimique
Elle est causée par la présence d’hétérogénéités dans le métal ou dans le réactif. Les hétérogénéités du métal sont d’origine chimique ou cristallographique. Les premières sont dues à l’existence de différentes phases, les secondes à la juxtaposition de régions bien recristallisées et de zones écrouies dans le métal. Dans les deux cas, si le potentiel de corrosion au contact du réactif n’est pas le même en tous les points du métal, une pile électrochimique prend naissance avec passage d’un courant électrique. On dit alors qu’il existe dans le métal des piles locales formées de microanodes et de microcathodes. Les régions anodiques, c’est-à-dire celles dont le potentiel est le plus négatif, sont le siège d’une attaque et des ions métalliques passent dans la solution.
Le fer contenant des inclusions de sulfure de fer, plongé dans un électrolyte, est ainsi le siège d’un phénomène de corrosion électrochimique. Ce sulfure de fer constitue l’anode de la pile et il est attaqué. La vitesse de l’attaque augmente avec la surface de sulfure au contact du réactif, c’est-à-dire avec la teneur du fer en sulfure.
Le processus de corrosion qui apparaît lorsque deux métaux différents sont couplés est le même. Ainsi, dans le cas du couple cuivre-fer, le cuivre constitue la cathode et le fer, dont le potentiel par rapport à l’électrolyte est le plus négatif, joue le rôle d’anode et se trouve attaqué.
La corrosion par hétérogénéité du réactif peut se développer même lorsque le métal est d’un très haut degré de pureté, donc que le courant débité par les piles locales y est très faible. Il suffit pour cela qu’il existe dans l’électrolyte un gradient de concentration des ions en solution ou des gaz dissous, en particulier de l’oxygène. Dans ce dernier cas, on a une pile de corrosion fonctionnant par différence de concentration en oxygène dissous dans le réactif. C’est l’effet Evans. Ce dernier phénomène apparaît très souvent dans la pratique, par exemple aux «recoins» des structures métalliques, c’est-à-dire dans des régions où l’oxygène peut difficilement diffuser alors que le réactif entourant les régions voisines est saturé en oxygène.
L’interprétation de la corrosion électrochimique, quelle qu’en soit l’origine, fait intervenir l’existence simultanée d’une réaction d’oxydation du métal sur les microanodes et d’une réaction de réduction de l’hydrogène ou de l’oxygène sur les microcathodes. Ainsi, dans le cas du fer plongé dans une solution aqueuse, on observe l’oxydation du fer (FeFe2+ + 2 e-) aux anodes et, suivant le pH, la réduction de l’hydrogène (2 H+ + 2 e-2 H) ou de l’oxygène (O2 + 4 e- + 2 H2O4 OH-) aux cathodes. L’intensité du courant qui correspond à ces deux réactions est égale en valeur absolue, mais, en fait, c’est le courant relatif à la réaction anodique qui est le seul responsable du phénomène de corrosion.
Méthodes d’étude
Elles font appel à des techniques très variées. L’examen visuel ou micrographique permet bien souvent de déceler le début du phénomène de corrosion, par exemple l’apparition de piqûres, ou de mettre en évidence sa morphologie.
Les méthodes pondérales ont pour but de mesurer la perte de poids des échantillons métalliques immergés dans le réactif pendant un temps donné. Si la corrosion est uniforme, elles conduisent à la détermination de la perte d’épaisseur par an. Elles peuvent être employées dans les milieux naturels et nécessitent alors un temps très long, par exemple plusieurs années, ou bien au laboratoire en essais accélérés par immersions et émersions alternées, et ne durent alors que quelques semaines. Dans certains cas, les échantillons sont placés sous contrainte afin de reproduire aussi exactement que possible les conditions réelles d’emploi.
Les isotopes radioactifs sont souvent utilisés avec succès en les introduisant soit dans le métal pendant son élaboration, de façon à suivre leur élimination progressive, soit dans la solution corrosive, afin de mettre en évidence la pénétration dans le métal d’un élément donné pendant que se développe le phénomène de corrosion.
Les méthodes électrochimiques sont très utilisées au laboratoire et même dans les installations industrielles. Elles permettent de déterminer par exemple le potentiel de corrosion d’un échantillon ou même d’une phase déterminée, et aussi de calculer l’intensité du courant responsable de la corrosion, donc, par application de la loi de Faraday, la perte de poids en un temps donné. Bien que leur utilisation ne puisse pas se faire dans tous les cas, elles rendent de grands services car elles sont rapides et permettent de façon simple d’effectuer des essais comparatifs, très utiles en particulier pour le choix du matériau à utiliser dans des conditions d’emploi données.
Moyens de lutte contre la corrosion
Trois méthodes principales sont utilisées: la première consiste à choisir un métal ou un alliage qui n’est pas attaqué ou peu attaqué par le milieu extérieur, la deuxième à modifier très légèrement la composition du réactif par addition de faibles quantités de corps appelés inhibiteurs; enfin, la troisième, de caractère électrochimique, consiste à imposer au métal un potentiel qui lui permet de ne pas être attaqué par le réactif.
L’addition, même à faible teneur, d’un élément d’alliage améliore le plus souvent les caractéristiques mécaniques des trois grands métaux de base que sont le fer, l’aluminium et le cuivre, ainsi que leur résistance à la corrosion dans certains milieux. Dans le cas du fer, par exemple, l’addition de chrome, de nickel, de molybdène est utilisée pour la préparation de différentes classes d’aciers inoxydables.
Cependant, l’utilisation d’aciers alliés constitue une solution onéreuse, et, dans bien des cas, une autre méthode peut être utilisée qui consiste, par exemple, à construire l’appareillage en acier ordinaire et à le protéger par un revêtement d’un métal ou d’un alliage résistant bien au milieu corrosif, et de faible épaisseur. On emploie ainsi des revêtements de zinc, d’étain, de nickel, de chrome, d’acier inoxydable, de métaux précieux.
Les inhibiteurs de corrosion sont ajoutés dans le réactif en très petite quantité, de l’ordre de 1 p. 1 000 en volume. Ils forment à la surface du métal un film continu et étanche qui l’isole du réactif. Leur nature est très variée: composés minéraux comme les phosphates et les chromates, ou composés organiques comme les alcools ou les amines. Ils sont utilisés en particulier pour la protection des échangeurs de chaleur, des unités pétrolières et aussi pour éviter l’attaque des métaux ferreux au cours du décapage acide.
La protection électrochimique des métaux constitue une solution très élégante; elle consiste à coupler la structure à protéger avec un métal moins noble, qui joue le rôle d’anode dans la pile ainsi formée. En utilisant des anodes de zinc ou de magnésium, qui sont détruites préférentiellement, on protège de manière courante les coques de navires, les installations portuaires ou les canalisations enterrées (fig. 3).
La protection cathodique est également réalisée à l’aide d’une source extérieure de courant qui permet de porter le potentiel du métal à une valeur suffisamment négative pour que la réaction d’oxydation n’ait pas lieu.
2. La corrosion sèche
La corrosion sèche correspond à l’attaque d’un métal M par un gaz G pour donner le composé C:

Tous les métaux et alliages, à l’exception des métaux nobles (l’or, par exemple), sont attaqués par les gaz à température suffisamment élevée. Parmi toutes les réactions possibles, ce sont surtout celles avec l’oxygène qui ont été le plus étudiées: au sens traditionnel du terme c’est l’oxydation. Mais, en fait, on peut donner à ce dernier terme son sens le plus large et qui caractérise alors l’action d’un élément électronégatif sur un métal ou un alliage: soufre, halogènes (Cl, Br, I), azote, etc. Ces différents éléments actifs peuvent être sous forme libre ou sous forme combinée: vapeur d’eau, monoxyde et dioxyde de carbone, anhydride sulfureux et hydrogène sulfuré, ammoniac, etc.
L’action de ces différents éléments obéit généralement aux mêmes principes et lois et s’étudie expérimentalement selon les mêmes méthodes. Il est alors possible de décrire plus particulièrement les réactions avec l’oxygène, étant bien entendu que les résultats peuvent être étendus à d’autres éléments électronégatifs.
On ne peut se limiter, pour comprendre les processus de l’oxydation, à appliquer seulement les lois de la thermodynamique. En effet, lorsqu’un oxyde se forme à la surface d’un métal, il isole de plus en plus celui-ci de l’oxygène; aussi, la vitesse de croissance de la couche d’oxyde n’est pas contrôlée par la réaction chimique (1) mais par la diffusion soit du métal, soit de l’oxygène à travers le film solide: c’est un processus physique.
Au cours de l’oxydation d’un métal, il est important de considérer le rapport du volume équivalent de l’oxyde formé à celui du métal attaqué (coefficient d’expansion):
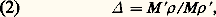
formule dans laquelle M et M sont les masses par équivalent-gramme et 福 et 福 , les masses volumiques de l’oxyde et du métal. Si est inférieur à 1, l’oxyde ne recouvre pas toute la surface et n’est donc pas protecteur: c’est le cas pour les métaux alcalins et alcalino-terreux. Si est supérieur à 1, l’oxyde est protecteur: c’est le cas pour les métaux usuels tels que le fer, le nickel et le cuivre.
En fait, les processus de l’oxydation sont relativement complexes, et leur interprétation nécessite de faire appel à différentes disciplines scientifiques. Les aspects structuraux et mécaniques, en particulier, revêtent une importance parfois prédominante.
Pour un métal donné, la vitesse d’attaque dépend de la température. Mais, en dehors de ce facteur important, d’autres paramètres interviennent directement sur le déroulement de la réaction: la pression partielle et la composition du gaz, la pureté du métal ainsi que sa structure et son état de surface.
Structure et défauts des oxydes. Diffusion
Pour que la réaction puisse se poursuivre, il est nécessaire que le métal et l’oxygène puissent venir au contact l’un de l’autre, c’est-à-dire qu’ils puissent diffuser à travers l’oxyde. Donc, suivant les cas, la réaction d’oxydation se produira: à l’interface métal-oxyde s’il s’agit d’une diffusion préférentielle d’oxygène (c’est le cas de Ti/Ti2, Zr/Zr2 et U/U2), à l’interface oxyde-oxygène s’il s’agit d’une diffusion préférentielle des ions métalliques (Fe/FeO et Ni/NiO).
Les oxydes sont des édifices cristallins dans lesquels l’oxygène et le métal sous forme ionique occupent des sites bien déterminés. Mais ces cristaux présentent des défauts ponctuels de deux types principaux: un cation interstitiel et une lacune de cation (défauts de Frenkel), comme pour AgBr; une lacune d’anion et une lacune de cation (défauts de Schottky), comme pour NaCl.
De plus, dans de nombreux cristaux, on observe un écart par rapport à la composition stœchiométrique. Ainsi, FeO et NiO, d’une part, ZnO, d’autre part, sont caractérisés respectivement par un défaut et par un excès de métal (fig. 4).
Ces imperfections du réseau cristallin sont à l’origine de la diffusion soit d’ions oxygène, soit d’ions métal dans les oxydes, et permettent d’expliquer les caractéristiques électriques de ces derniers (conductibilité et nombre de transport). On conçoit alors aisément que la nature et la vitesse de migration des ions dépendront du type de défauts présents dans l’oxyde formé.
Lois cinétiques d’oxydation des métaux
Le paramètre important pour étudier quantitativement les phénomènes d’oxydation est la variation de l’épaisseur e de la couche d’oxyde ou la variation m du poids de l’échantillon en fonction du temps t pour une température et une pression données.
Plusieurs théories (Wagner, Mott et Cabrera, Hauffe et Ilschner) ont été proposées pour traduire quantitativement l’oxydation des métaux. Diverses lois cinétiques d’oxydation ont été observées (fig. 5).
Le type de loi dépend fortement de l’épaisseur du film d’oxyde; il dépend donc du temps et de la température. Les conditions d’observations sont schématiquement les suivantes:
– la loi linéaire est observée quand la cinétique est gouvernée par la réaction chimique; la masse volumique de l’oxyde étant supérieure à celle du métal, la couche d’oxyde ne recouvre pas la surface du métal d’une façon continue et l’oxygène peut toujours atteindre le métal pour l’oxyder;
– les lois cubique ou parabolique sont observées aux températures moyennes quand la couche d’oxyde est protectrice et mince; la température étant suffisamment basse, les ions diffusent très difficilement à travers le film d’oxyde; toutefois, les électrons du métal peuvent traverser l’oxyde quand il est encore très mince et venir réagir avec l’oxygène; il s’établit alors un champ électrique intense qui entraîne la migration des ions vers l’interface oxyde-oxygène;
– la loi parabolique est observée pour de nombreux métaux courants quand la couche d’oxyde est épaisse et protectrice; l’oxydation est assurée par la diffusion des ions à travers le film d’oxyde (loi de Wagner); contrairement au cas des couches minces, pour lesquelles le gradient de potentiel électrique joue le rôle principal, pour une couche épaisse c’est le gradient de concentration qui est essentiel; la constante parabolique k 2 est exprimée en fonction des coefficients de diffusion des ions à travers l’oxyde;
– la loi logarithmique ainsi que la loi logarithmique inverse sont observées à basse température pour les films minces d’oxyde; dans certains cas (silicium, aluminium, chrome), la loi devient asymptotique.
L’analyse des vitesses d’oxydation, résumée ci-dessus pour un métal pur, est valable quel que soit le nombre de degrés d’oxydation du métal considéré.
Processus de l’oxydation
Lorsqu’un métal est placé dans une atmosphère gazeuse, il y a adsorption du gaz à la surface selon un mode qui varie avec les conditions: aux basses températures, il y a adsorption physique, caractérisée par la fixation d’oxygène moléculaire grâce aux forces de Van der Waals; l’adsorption chimique, qui se produit aux températures intermédiaires et élevées, est caractérisée par la fixation d’atomes ou d’ions à la surface du métal avec apparition de liaisons fortes de type ionique.
Dans des conditions d’oxydation très ménagées, l’oxyde se forme en des sites particuliers de la surface du métal comme cela a été montré sur le fer; c’est la germination. Trois étapes successives ont été distinguées dans ce processus (germination et croissance): la période d’incubation , pendant laquelle s’édifie un film primaire polycristallin constitué de microcristaux orientés au hasard; à la fin de cette période, il apparaît brusquement un certain nombre de germes (fig. 6) qui sont monocristallins et dont la morphologie et le nombre dépendent de l’orientation cristallographique du métal; leur nombre augmente quand la température diminue et quand la pression augmente; la période de croissance latérale des germes, qui finissent par recouvrir toute la surface; enfin, la période de croissance uniforme de l’oxyde.
Deux types d’explication ont été proposés pour interpréter les causes de la localisation des germes: la germination «hétérogène», dans laquelle la vitesse de réaction est accélérée au voisinage de défauts physiques ou chimiques (la sulfuration du cuivre fournit un exemple en faveur de cette interprétation); la germination «homogène», observée aux températures élevées pour les métaux ayant peu de défauts.
Une relation particulière d’orientation (épitaxie) associée à une plus grande perfection du réseau favorise le développement préférentiel des cristallites du film primaire qui possèdent cette orientation.
Après l’adsorption irréversible, il se forme, à la surface du métal, un film d’oxyde dont l’épaisseur croît avec le temps. Quand celle-ci est comprise entre quelques centièmes et quelques dixièmes de micromètre, on observe des colorations dues à un phénomène d’interférences des rayons lumineux réfléchis à la surface de l’oxyde et à l’interface métal-oxyde. L’épaisseur du film dépend de l’orientation cristallographique du métal: c’est la croissance épitaxique (fig. 7).
Lorsque l’épaisseur de la couche d’oxyde devient supérieure à 1 micromètre environ, l’évolution de la corrosion est différente de celle qui a été envisagée précédemment. La vitesse de croissance de l’oxyde dépend de la diffusion à travers celui-ci, diffusion qui assure le contact entre le gaz corrosif et le métal. L’oxydation tend alors dans bien des cas à devenir indépendante des réactions aux interfaces. Suivant que le métal possède un seul ou plusieurs degrés d’oxydation, l’oxyde est composé d’une couche unique ou de plusieurs couches superposées (fig. 8). Très souvent, conformément à la théorie, une loi de croissance parabolique est observée. La constante de vitesse k augmente avec la température suivant la loi:

Q étant l’énergie d’activation du phénomène.
De nombreux paramètres, en particulier les caractéristiques mécaniques relatives du métal et de l’oxyde, affectent la morphologie des pellicules épaisses. Celles-ci présentent dans bien des cas des défauts: fissures, pores, cavités, décollements et pustules. Ces défauts qui perturbent les mécanismes de croissance entraînent alors des modifications importantes, comme celles que l’on rencontre en pratique dans les couches de calamines, qui sont assez différentes des couches idéales.
Corrosion des alliages
D’un point de vue pratique, de nombreux alliages présentant à la fois de bonnes propriétés mécaniques et une bonne résistance à la corrosion ont été développés. Il serait très satisfaisant de pouvoir prévoir les effets d’une addition métallique sur les caractéristiques de la tenue à la corrosion d’un alliage dans des conditions données. Dans le cas de l’oxydation, par exemple, on peut évaluer l’augmentation ou la diminution du nombre de défauts lorsqu’on introduit un ion étranger dans l’oxyde, ce qui correspond à une accélération ou à un ralentissement de l’oxydation (théorie de Wagner-Hauffe). Mais, dans bien des cas, une telle théorie ne s’applique pas. En effet, le problème de la corrosion sèche des alliages est très complexe et très délicat car, en plus des phénomènes décrits précédemment dans le cas d’un métal pur, de nouveaux paramètres sont à prendre en considération: les affinités respectives des éléments de l’alliage à l’égard des gaz; les vitesses de diffusion des éléments d’alliage dans le produit de corrosion et la vitesse de diffusion du gaz dans l’alliage; la solubilité des éléments d’alliage dans la couche corrodée.
Protection contre la corrosion sèche
Pour lutter efficacement contre la corrosion sèche, on peut utiliser des revêtements métalliques. Le revêtement idéal devrait avoir de nombreuses caractéristiques: être lui-même inactif à l’égard des gaz; avoir une bonne ductilité; avoir une bonne adhérence insensible aux variations de température; ne pas former par diffusion avec le substrat une sous-couche fragile; se cicatriser en cas de détérioration accidentelle; résister à l’érosion.
Aucun revêtement ne présentant à la fois toutes ces qualités, il est nécessaire de trouver une solution de compromis. Divers procédés ont été mis au point parmi lesquels on peut citer à titre d’exemples: l’immersion dans un métal liquide (aluminisation); le dépôt électrolytique suivi d’une diffusion thermique (Ni et Cr); la cémentation en phase gazeuse (Cr ou Si sur le fer); la projection à chaud, ou schoopage (Al, Ni); le procédé thermochimique Galmiche-O.N.E.R.A. (chromaluminisation et tantalisation des superalliages réfractaires pour aubes de turbines à gaz aéronautiques).
Les matériaux céramiques (oxydes, nitrures, carbures) sont également utilisés comme revêtements. Ceux-ci s’obtiennent soit par projection à chaud (en particulier à l’aide du chalumeau à plasma), soit par oxydation d’un dépôt métallique, soit encore par oxydation d’un des éléments de l’alliage. Ainsi, dans le cas des aciers inoxydables, le chrome s’oxyde préférentiellement pour former une couche protectrice constituée de Cr23.
corrosion [ kɔrozjɔ̃ ] n. f.
• v. 1300; lat. corrosio
♦ Action de corroder; son résultat. ⇒ brûlure, désagrégation, destruction, 2. usure; rouille. Corrosion par un acide. — Géol. Dissolution produite par les eaux de ruissellement. ⇒ érosion, ravinement; karst.
● corrosion nom féminin (bas latin corrosio, -onis, action de ronger) Action de corroder ; fait d'être corrodé. Dégradation d'un matériau sous l'action du milieu ambiant et par un processus autre que mécanique. ● corrosion (expressions) nom féminin (bas latin corrosio, -onis, action de ronger) Figures de corrosion, figures qui apparaissent à la surface d'un cristal lorsqu'il est attaqué par un réactif ou un solvant approprié. (Elles révèlent l'anisotropie et les symétries du cristal et sont dues à la présence de défauts internes ou superficiels.) ● corrosion (synonymes) nom féminin (bas latin corrosio, -onis, action de ronger) Figures de corrosion
Synonymes :
- piqûres d'attaque
corrosion
n. f. Action ou effet de ce qui est corrosif.
|| CHIM Détérioration superficielle des métaux d'origine chimique ou électrochimique (partic. sous l'effet de l'humidité, du sel, etc.). La corrosion du fer par l'acide.
|| GEOL Corrosion des sols, par les eaux de ruissellement.
⇒CORROSION, subst. fém.
A.— Action de corroder (cf. corroder A); résultat de cette action. Agent de corrosion. (Quasi-)synon. désagrégation, destruction, usure. Pour diminuer les corrosions on a proposé de mettre des plaques de zinc dans les eaux capables d'attaquer le fer (BOULANGER, Malterie, brasserie, 1934, p. 34). La corrosion annuelle des falaises est parfois considérable (QUINETTE DE ROCHEMONT, Trav. mar., 1900, p. 92).
SYNT. Corrosion bio-chimique, biologique; corrosion intense, lente, progressive; corrosion d'un acide, du fer, des rives, d'un tuyau de gaz; être atteint de corrosion; protéger contre la corrosion, provoquer des corrosions, résister à la corrosion.
— Emplois spéc.
1. CRISTALLOGR. Figure de corrosion. ,,Modification de la surface d'un cristal quand on attaque, à l'aide d'un réactif approprié, une face naturelle ou une face de clivage`` (DUVAL 1959).
2. ODONTOLOGIE. ,,Altération des tissus durs de la dent, secondaire à l'action d'un acide, d'origine professionnelle, alimentaire ou médicamenteuse`` (COURTOIS 1972). ,,Altération lente de la surface d'une obturation métallique (amalgame ou inlay) par la salive ou les médicaments`` (COURTOIS 1972).
B.— Au fig. (cf. corroder B), rare. La corrosion des partis par la guerre (PROUDHON, Révol. soc., 1852, p. 159) :
• Pendant trente ans ils se sont mis sur le pied de ruiner tout ce qui était debout en France (...). Et aujourd'hui nous n'aurions pas le droit de ruiner cette ruine. Nous n'aurions pas le droit de corroder cette corrosion, d'éroder cette érosion.
PÉGUY, L'Argent, 1913, p. 1199.
Prononc. et Orth. :[ (R)R
(R)R ]; pour [
]; pour [ ] ou [RR] cf. corrosif. Admis ds Ac. 1694-1932. Étymol. et Hist. Ca 1300 « action de corroder; état de ce qui est corrodé » (La Chirurgie de l'abbé Poutrel, 27 v° 2 ds Mélanges Lecoy, 1973, p. 543); 1756 fig. (MIRABEAU [Marquis de], L'Ami des hommes, I, 57 ds GOHIN, p. 343 : une corrosion continuelle et croissante du nerf de la population). Empr. au b. lat. corrosio « action de ronger; morsure ». Fréq. abs. littér. :6. Bbg. GOHIN 1903, p. 343. — Termes techn. fr. Paris, 1972, p. 12.
] ou [RR] cf. corrosif. Admis ds Ac. 1694-1932. Étymol. et Hist. Ca 1300 « action de corroder; état de ce qui est corrodé » (La Chirurgie de l'abbé Poutrel, 27 v° 2 ds Mélanges Lecoy, 1973, p. 543); 1756 fig. (MIRABEAU [Marquis de], L'Ami des hommes, I, 57 ds GOHIN, p. 343 : une corrosion continuelle et croissante du nerf de la population). Empr. au b. lat. corrosio « action de ronger; morsure ». Fréq. abs. littér. :6. Bbg. GOHIN 1903, p. 343. — Termes techn. fr. Paris, 1972, p. 12.
(R)R]; pour [] ou [RR] cf. corrosif. Admis ds Ac. 1694-1932. Étymol. et Hist. Ca 1300 « action de corroder; état de ce qui est corrodé » (La Chirurgie de l'abbé Poutrel, 27 v° 2 ds Mélanges Lecoy, 1973, p. 543); 1756 fig. (MIRABEAU [Marquis de], L'Ami des hommes, I, 57 ds GOHIN, p. 343 : une corrosion continuelle et croissante du nerf de la population). Empr. au b. lat. corrosio « action de ronger; morsure ». Fréq. abs. littér. :6. Bbg. GOHIN 1903, p. 343. — Termes techn. fr. Paris, 1972, p. 12.
corrosion [kɔʀozjɔ̃] n. f.
ÉTYM. V. 1300; bas lat. corrosio « action de ronger, morsure », du supin de corrodere. → Corroder, corrosif.
❖
1 (V. 1300). Action de corroder; résultat de cette action. ⇒ Brûlure, désagrégation, destruction, usure. || La corrosion d'une substance par un acide. || Agent de corrosion. || Résister à la corrosion. || Corrosion de contact : usure de contact par réaction chimique (⇒ Abrasion). — Mar. || Corrosion de l'eau de mer sur la coque d'un navire.
1 (…) les navires de commerce opposent à la corrosion l'épaisseur de leurs tôles. C'est vrai : une tôle de 15 à 20 mm (navire) résistera plus longtemps que les tôles de 5 et 6 mm employées dans la construction des yachts.
Bernard Moitessier, Cap Horn à la voile, p. 42.
♦ Géol. Dissolution produite par les eaux de ruissellement. ⇒ Érosion, ravinement.
♦ Techn. || Figure de corrosion : modification de la surface d'un cristal attaqué par un réactif donné.
♦ Chir. dent. Altération des dents, de la surface d'une obturation.
2 (1756). Le fait d'être attaqué (la corrosion d'un sentiment, etc.); le fait d'attaquer, de ronger. ⇒ Corroder, 2.
2 Il entrevoit une corrosion qui s'amorce, puis qui se propage, comme la flamme, tant qu'il lui reste un aliment (…)
J. Romains, les Hommes de bonne volonté, t. III, V, p. 86.
Encyclopédie Universelle. 2012.