- acquired h. nonfamilial h. that develops as a consequence of some primary disease, such as thyroid deficiency.
- familial h. a group of diseases characterized by changes in concentration of β-lipoproteins and pre-β-lipoproteins and the lipids associated with them. See type I familial h., type II familial h., type III familial h., type IV familial h., type V familial h..
- lipoprotein(a) h. elevated levels of lipoprotein(a) in the serum; associated with an increased risk of coronary disease.
- type I familial h. [MIM*238600] h. characterized by the presence of large amounts of chylomicrons and triglycerides in the plasma when the patient has a normal diet, and their disappearance on a fat-free diet; low α- and β-lipoproteins on a normal diet, with increase on a fat-free diet; decreased plasma postheparin lipolytic activity; and low tissue lipoprotein lipase activity. It is accompanied by bouts of abdominal pain, hepatosplenomegaly, pancreatitis, and eruptive xanthomas; autosomal recessive inheritance; caused by mutation in the lipoprotein lipase gene (LPL) on chromosome 8p. SEE ALSO: familial lipoprotein lipase inhibitor. SYN: Bürger-Grütz syndrome, familial fat-induced hyperlipemia, familial hyperchylomicronemia, familial hypertriglyceridemia (1), idiopathic hyperlipemia.
- type II familial h. [MIM*143890 and MIM*144400] h. characterized by increased plasma levels of β-lipoproteins and cholesterol, elevated or normal levels of triglycerides; heterozygotes have mild lipid changes and are susceptible to atherosclerosis in middle age, but homozygotes have severe changes—often with generalized xanthomatosis, xanthelesma, corneal arcus, and frank clinical atherosclerosis as young adults. This disorder is divided into two classes, both inherited as autosomal dominant with homozygotes more severely affected than heterozygotes: 1) type IIA, which is characterized by elevated LDL but normal triglycerides and is due to a deficiency of the LDL receptor, a defect of the receptor or a modified LDL-apolipoprotein B-100, caused by mutation in the LDL receptor (LDLR) gene on chromosome 19p. SYN familial hypercholesterolemia; 2) type IIB has elevated LDL, cholesterol, and triglycerides, due to dysregulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG CoA reductase), the rate-controlling enzyme in cholesterol biosynthesis. SYN familial hyperbetalipoproteinemia, familial hypercholesterolemic xanthomatosis.
- type III familial h. [MIM*107741] h. characterized by increased plasma levels of LDL, β-lipoproteins, pre-β-lipoproteins, cholesterol, phospholipids, and triglycerides; hypertriglyceridemia induced by a high carbohydrate diet, and glucose tolerance is abnormal; frequent eruptive xanthomas and atheromatosis, particularly coronary artery disease; biochemical defect lies in apolipoproteins; there are many varieties; one variety is caused by mutation in the APOE gene on chromosome 19q. SYN: carbohydrate-induced hyperlipemia, dysbetalipoproteinemia, familial hyperbetalipoproteinemia and hyperprebetalipoproteinemia, familial hypercholesterolemia with hyperlipemia.
- type IV familial h. [MIM*144600] plasma levels of VLDL, pre-β-lipoproteins and triglycerides are increased on a normal diet, but β-lipoproteins, cholesterol, and phospholipids are normal; hypertriglyceridemia is induced by a high carbohydrate diet; may be accompanied by abnormal glucose tolerance and susceptibility to ischemic heart disease; probably autosomal dominant inheritance but genetic heterogeneity is a possibility. SYN: carbohydrate-induced hyperlipemia, familial hyperprebetalipoproteinemia, familial hypertriglyceridemia (2).
- type V familial h. [MIM*144650] h. characterized by increased plasma levels of chylomicrons, VLDL, pre-β-lipoproteins, and triglycerides, and slight rise of cholesterol on a normal diet, with β-lipoproteins normal; may be accompanied by bouts of abdominal pain, hepatosplenomegaly, susceptibility to atherosclerosis, and abnormal glucose tolerance; probably autosomal recessive inheritance. SYN: combined fat- and carbohydrate-induced hyperlipemia, familial hyperchylomicronemia with hyperprebetalipoproteinemia, mixed hyperlipemia.
* * *
hy·per·li·po·pro·tein·emia or chiefly Brit hy·per·li·po·pro·tein·ae·mia -.lī-pə-.prō-tē-'nē-mē-ə, -.lip-ə-, -.prō-tē-ə-'nē- n the presence of excess lipoprotein in the blood
* * *
hy·per·lipo·pro·tein·emia (hi″pər-lip″o-pro″te-neґme-ə) an excess of lipoproteins in the blood, due to a disorder of lipoprotein metabolism; it may be an acquired or familial condition or some combination. The disorder has been subdivided on the basis of biochemical phenotype, and each type has since been shown to have a variety of causes. See accompanying table for individual phenotypes as well as some primary genetic disorders that cause them. See also familial h., hyperlipemia, and hyperlipidemia.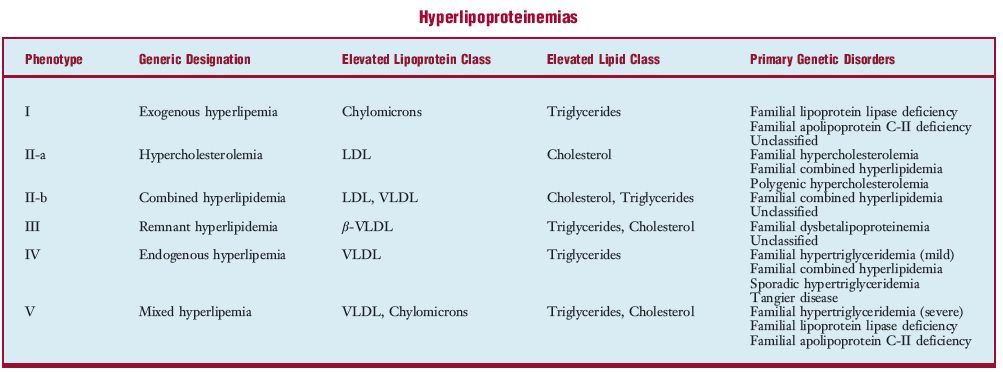
LDL, Low-density lipoproteins; VLDL, very-low-density lipoproteins; b-VLDL, a class of abnormal VLDL.
Medical dictionary. 2011.